To clot or not to clot To clot









![Warfarin Resistance • Day 1: – Frequent sampling to: • Ascertain peak [warfarin] •](https://slidetodoc.com/presentation_image_h/2cae96f0176a4f344d31345575cb7578/image-10.jpg "Warfarin Resistance • Day 1: – Frequent sampling to: • Ascertain peak [warfarin] •")

 • Adopted in 1983 as the WHO international PT standardisation")
. This must be")
/GMNPT (s))ISI Thus to generate accurate INRs it")



 • First described in 1926 By Dr Eric Adolf Von.")
 • VWD patients tend to bleed from: – Mucosal surfaces")
 • Functions: 1. Attach to subendothelial collagen and to platelets,")
 • Phenotypes: – Type 1 • Mild quantitative VWF deficiency.")
 • Can be caused by: – Specific or non-specific antibodies –")
")


, Factor")
 2. Moderate 1")



 • Neonate/Toddler Swelling at site of internal bleeding Warm at site")










 Longterm health conditions")
 – Time taken (sec) for blood being")

 – Time taken (sec)")




 which extends half-life – More expensive")
")
 Monoclonal Ab for treatment of Haemophilia A with, or without, inhibitors")
")





- Slides: 60
To clot, or not to clot?
To clot?
Anticoagulant Therapy Patients who have had, or are at risk from developing a thrombosis (clot) are treated with anticoagulant drugs. Two most common anticoagulant drugs: 1. Warfarin 2. Unfractionated heparin
Risks of anticoagulant therapy 1. Bleeding – – • • May happen even if treatment is within therapeutic range Can be reversed with oral or Intravenous vit. K Dose should reflect INR and severity of bleeding If bleeding is life threatening then immediate reversal of anticoagulation can be achieved with concentrates of FII, FVII and FX. If these are not available use fresh frozen plasma (FFP) 2. Warfarin-induced skin necrosis – • – – Rare, but serious Usually presents in first 3 -5 days of therapy Higher incidence in patients with thrombophilia defect (esp. Protein C deficiency). Formation of microthrombi in the skin which reduces O 2 supply to surrounding tissues, and eventually cell death
Anticoagulant Therapy is a delicate balancing act between: Hypocoagulability and Hypercoagulability Overdose = bleeding Underdose = inadequate prevention of thrombosis
Warfarin • Interferes with the synthesis of vitamin Kdependent factors by diminishing their biological activity – FII – FVII – FIX – FX • Also interferes with production of: – Protein C – Protein S (a co-factor of protein C) – Protein Z
Warfarin • Regularly monitored using PT – Because most factors that contribute to the clotting time are vit. K-dependent! • PT needs to be converted to a ratio called the International Normalized Ratio (INR) Approximately 1% of UK population receive warfarin therapy at any given time. Thus PT/INR monitoring is a large part of routine haemostasis lab work
Warfarin The response to a given dose of warfarin varies from patient to patient – hence the need for regular INR monitoring Some patients demonstrate a poor response to warfarin, even at increasingly higher doses This can be due to warfarin resistance
Warfarin Resistance Supervised oral warfarin dose • What at first may appear to be warfarin resistance may be due to non-compliance of the patient in taking their medication – hence supervision is required Serial blood samples then taken over 3 days 1. To determine INR • Helps determine: – Pharmacodynamic response 2. To directly assay warfarin – bioavailability of warfarin – Warfarin half-life
Warfarin Resistance • Day 1: – Frequent sampling to: • Ascertain peak [warfarin] • Assess degree of absorption • Day 2: – Sample taken at 48 hours post-dose • Day 3: – Sample taken at 72 hours post-dose These samples help ascertain the half-life of warfarin
Warfarin Resistance Rarely, the results may suggest warfarin malabsorption Requires further study by a specialist laboratory: Intravenous warfarin administration to assess warfarin resistance post-absorption
International Normalised Ratio (INR) • Adopted in 1983 as the WHO international PT standardisation scheme • Derived from the ratio between time taken for blood to clot normally compared to time taken for blood to clot due to warfarin – Thus: • If normal PT = 14 seconds • Warfarin PT = 28 seconds • INR = 28/14 = 2. 0 However…
INR Calculation of PT ratio (patient PT / mean normal PT). This must be from geometric mean normal prothrombin time (GMNPT) for the thromboplastin in use GMNPT determination: – Should be derived locally to generate accurate INR results from a minimum of 20 fresh plasmas from normal adults (both ♂ and ♀) – Preferred to arithmetic mean as there is a log-normal distribution of PTs in a healthy adult population. • Thus calculating GMNPT is effectively performing a log transformation to produce a normal distribution prior to calculating the mean Sensitivity of the thromboplastin to oral anticoagulant effect relative to WHO standard is defined by the International Sensitivity Index (ISI) • ISIs are assigned to thromboplastins after calibration against WHO standards, or a secondary standard calibrated against a WHO standard INR is therefore calculated from the PT ratio and ISI like this…
INR Calculation: INR = (Patient PT (s)/GMNPT (s))ISI Thus to generate accurate INRs it is essential for the ISI assignment to also be accurate. From our previous example… • If normal PT = 14 seconds • Warfarin PT (GMNPT) = 28 seconds • INR = 28/14 = 2. 0 If ISI = 1, then INR remains 2. 0 BUT… If ISI = 1. 4, then INR = (28/14)1. 4 = 2. 64
INR/ISI System • The INR/ISI system means patients results are equivalent regardless of the reagents or analytical platform used to generate the PT and prevents unnecessary and potentially dangerous alterations in prescribed dose of anticoagulant
Patient risk of thrombosis Low risk of thrombosis? : – Prescribed warfarin so INR = 2. 0 – 3. 0 High risk of thrombosis? : – Prescribed warfarin so INR = 3. 0 – 4. 0
Not to clot?
von Willebrands Disease (VWD) • First described in 1926 By Dr Eric Adolf Von. Willebrand (originally termed “pseudo-haemophilia”) • Autosomal inheritance • Dysfunction of Von Willebrand Factor – – VWF made in endothelial cells and secreted into plasma Also made in megakaryocytes Found in platelets Whilst still in cell units of VWF form huge multimers (HMW multimers)
von Willebrands Disease (VWD) • VWD patients tend to bleed from: – Mucosal surfaces (e. g. nose, mouth) – Small cuts – GI tract – Females often have menorrhagia and bleeding with miscarriage – VWF levels may rise in pregnancy
Von. Willebrands Factor (VWF) • Functions: 1. Attach to subendothelial collagen and to platelets, promoting formation of a platelet plug at the site of injury to small vessels 2. Bind to and transport FVIII • Thus if v. WF levels are reduced, levels of FVIII will also be reduced!
Von. Willebrands Disease (VWD) • Phenotypes: – Type 1 • Mild quantitative VWF deficiency. Deficiency of VWF due to reduced production, defective release or increased clearance – Type 2 variants are qualitative deficiencies • Type 2 A – Reduced platelet dependent function due to absence of high molecular weight (HMW) multimers • Type 2 B – Increased affinity to platelets. HMW multimers are absent, may show platelet deficiency. FVIII unaffected • Type 2 M – Decreased VWF-dependent platelet adhesion due to dysfunctional HMW multimers • Type 2 N – Normal VWF-dependent platelet function, but VWF has reduced affinity for FVIII – Type 3 • Severe quantitative bleeding disorder due to markedly reduced or absent HMW multimers and markedly reduced FVIII (~2%) – Pseudo VWD • Platelets have increased affinity for HMW multimers. Multimers and platelets constantly removed from circulation. Treated with platelet infusions. Dominant inheritance
Acquired VWD (AVWD) • Can be caused by: – Specific or non-specific antibodies – Adsorption of VWF onto malignant cell clones – Hypothyroidism – Loss of high molecular weight multimers under conditions of high shear stress (e. g. aortic stenosis)
Haemophilia: The Royal Disease Waldemar of Prussia Died aged 56 Henry (brother of Waldemar) Died aged 4 Frederick William Died aged 3 Czarevitch Alexis Romonov Murdered Viscount Trematon Died aged 20 Alfonso & Gonzalo (brothers) Both bled to death after an accident
Alex Dowsett
Chris Bombardier First Haemophiliac to reach the summit of Mount Everest (22 nd May 2017)
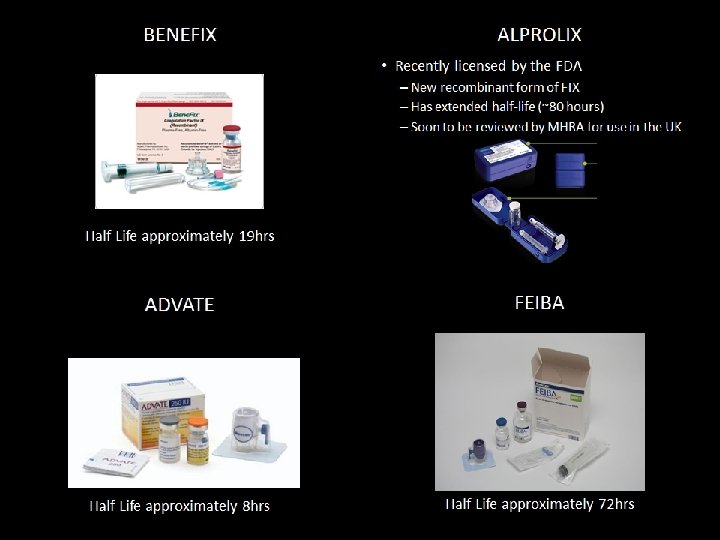
Haemophilia X-Linked Recessive inheritance Haemophilia A (Classic Haemophilia, 1: 10, 000 live births), Factor VIII deficiency • Treated with: ADVATE (mainly, there are other medicines, such as Kogenate, Alphanate, and so forth, but the most commonly used is ADVATE. Eloctate is the main extended half-life clotting factor currently in use) Haemophilia B (Christmas Disease, 1: 50, 000 live births), Factor IX deficiency • Treated with: BENEFIX (mainly, though several new extended half-life treatments, such as Idelvion/Alprolix, are now and soon will become available, but with attached conditions from NHS England) Autosomal inheritance Haemophilia C, Factor XI deficiency • • • Treated with FXI concentrate, FFP, Fibrin glue, antifibrinolytic drugs Higher incidence (8%) in Ashkenazi Jews (Eastern European descent) Few, or no symptoms (no bleeding into joints or muscles) Relationship between circulating FXI levels and bleeding severity is unclear Most common of the rare bleeding disorders (second most common in women, after VWD) Autoimmune response Acquired Haemophilia • Treated with FEIBA (activated prothrombin complex concentrate - APCC) • Tranexamic acid (Cyclo. Kapron) – antifibrinolytic drug esp. useful for mucous membranes (NOT to be used with APCC’s)
Phenotypes: 1. Mild 5 -50% (0. 05 – 0. 5 IU/ml) 2. Moderate 1 -5% (0. 01 – 0. 05 IU/ml) 3. Severe <1% (<0. 01 IU/ml)
Specialist Treatment & Care Specialist treatment centres are located throughout the UK Comprehensive Care Centres (CCCs) provide specialist support in all aspects of health of patients with other bleeding disorders: • • • Factor replacement (not available from GP/Community Pharmacy) Home therapy Home Visits (when required) Genetic Screening/Counselling Dentistry Orthopaedics Liason with other medical/surgical specialties Liason with schools HIV, HCV, etc Physiotherapy Clinical Psychology & Counselling Social Workers
Bleeding Episodes External bleeding: – Requires immediate attention and factor replacement as soon as possible – Easier to visualise what is wrong and how to stem bleeding using conventional methods mixed with factor replacement (and/or tranexamic acid if appropriate ) – Depending on age, severity, site, experience and advice from haemophilia centre staff, may require immediate attention at comprehensive care centre
Bleeding Episodes Internal bleeding: – Most dangerous form – Extent of bleeding not always immediately evident – Requires immediate treatment to prevent bleeding from progressing or getting worse – Depending on age, severity, site, experience and advice from haemophilia centre staff, may require immediate attention at comprehensive care centre
Symptoms (internal bleeding) • Neonate/Toddler Swelling at site of internal bleeding Warm at site of internal bleeding Painful to touch/move If bleeding is severe, may develop symptoms of hypovolemic shock (pallor, clammy, etc. ) – Continued crying/screaming for no apparent reason – – • Paediatric/Adult – As above, but able to explain what they are feeling and where – Pain often described as “pressure” or “resistance” – Haematoma may, or may not, be visible
Internal Bleeding – Left Knee
Epistaxis A. K. A. “nose bleeds” – Less common in haemophilia than internal injuries, but does happen NEVER hold head back ALWAYS lean head forward Pinch soft part of nose firmly until bleeding has ceased If appropriate, topical administration of tranexamic acid may be beneficial • Treat with factor as soon as possible • If neonate/toddler/young child seek advice from comprehensive care centre staff • •
Global Health Inequalities & Life Expectancy Varies depending on availability/receipt of proper treatment. Without adequate treatment, many people with haemophilia die before they reach adulthood. With proper treatment, life expectancy is about 10 years less than average, and children can look forward to a normal life expectancy.
Haemophilia Carriers Phenotypes: – – Normal Mild Moderate Severe “Low-level” carrier - thus will exhibit symptoms of haemophilia and require treatment Lyonization (or X-inactivation): – In females, the phenomenon by which one X chromosome (either maternally or paternally derived) is randomly inactivated in early embryonic cells, with fixed inactivation in all descendant cells – First described by the geneticist Mary Lyon – The repression of one of the two X-chromosomes in the somatic cells of females as a method of dosage compensation; at an early embryonic stage in the normal female, one of the two Xchromosomes undergoes inactivation, apparently at random, from this point on all descendent cells will have the same X-chromosome inactivated as the cell from which they arose, thus a female is a mosaic composed of two types of cells, one which expresses only the paternal X-chromosome, and another which expresses only the maternal X-chromosome.
Secondary Complications • Inhibitors • Haemophillic Arthropathy – Generally in weight bearing joints – Potentially leading to prosthetic joint replacement or fusion (Knee, Hip, Elbow, Ankle) • • • HIV HBV, HCV, HGV Liver Failure v. CJD Depression
Inhibitors • Most common is FVIII – – Approx’ 30% severe haemophilia A patients Alloantibodies against FVIII replacement Antibody: FVIII ratio = 1: 1 8% moderate/mild haemophilia A • Autoantibodies • Rapid FVIII inactivation followed by dissociation. Thus residual FVIII activity remains • Can also occur in non-haemophiliacs – “acquired inhibitors” • 12% Severe haemophilia B patients develop inhibitors with overall incidence of approximately 3%
Inhibitors • The type of gene mutation is a factor in the development of inhibitors – EG gene deletions, nonesense mutations – These mutations are less common in haemophilia B, hence inhibitors is less common in this phenotype • Site of mutation is also a factor which influences inhibitor formation
Inhibitors • Treatment: – High dose factor concentrate – Factor VIII bypassing agents: • r. FVII – Bypasses FVIII in clotting cascade • FEIBA (activated prothrombin complex concentrate - APCC) – Tranexamic acid (Cyclo. Kapron) – antifibrinolytic drug esp. useful for mucous membranes (NOT to be used with APCC’s) – Epsilon aminocaproic acid (AMICAR™) is an antifibrinolytic drug that can be given as an additional therapy in pill form or by injection to help hold clots in place in certain parts of the body, such as the mouth, bladder, and uterus.
Inhibitors • Treatment: – Plasmapheresis is a procedure that removes inhibitors from the person's bloodstream. It is usually done when the inhibitor titer needs to be brought down quickly (for example, before major surgery or in cases of severe bleeding that are not well controlled with bypassing agents). – Immune Tolerance Induction (ITI) therapy involves giving the person with inhibitors frequent doses of factor concentrates over several months, or sometimes years, to train the body to recognize the treatment product without reacting to it. This process is called tolerance induction. If a person plans to undergo immune tolerance induction therapy, but has not yet started, it is better not to use factor products to treat acute bleeding episodes because they are likely to provoke a rise in inhibitor titer.
Haemophillic Arthropathy
Depression Common to most (if not all) Longterm health conditions
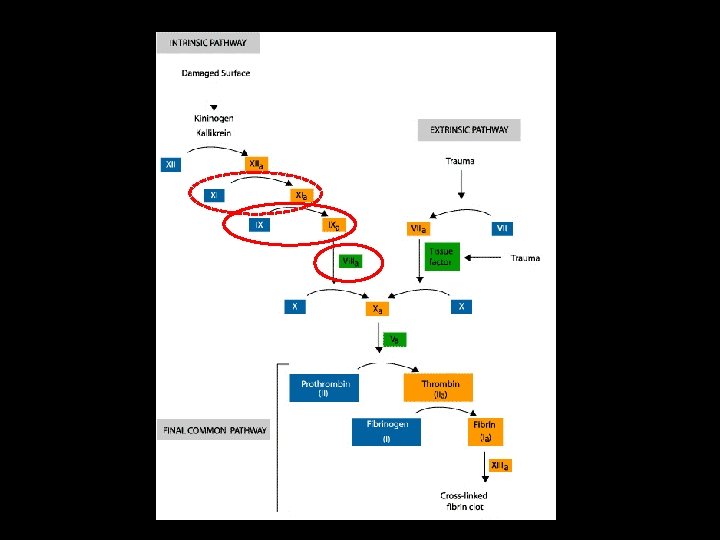
Relevant Laboratory Investigations • Prothrombin Time (PT) – Time taken (sec) for blood being tested to form a clot from the extrinsic and common pathways, with the result being referred to a reference range (which will vary depending on the techniques and type of thromboplastin used). Warfarin therapy = Raised PT Haemophilia = Normal PT v. WD = Normal PT
Relevant Laboratory Investigations • Raised PT could indicate deficiencies of: – – – FII FV FVII FX Fibrinogen Autoantibodies (Inhibitors) against any of the above clotting factors Anticoagulant drugs (e. g. Warfarin) affecting Vit. K-dependant factor production, or directly affecting thrombin (e. g. hirudin) Vit. K deficiency Liver disease Disseminated Intravascular Coagulation (DIC) Lupus anticoagulants (rare)
Relevant Laboratory Investigations • Activated Partial Thrombosplastin Time (a. PTT) – Time taken (sec) for blood being tested to form a clot from the intrinsic and common pathways, with the result being referred to a reference range. Warfarin therapy = Raised a. PTT Haemophilia = Raised a. PTT v. WD = Raised a. PTT
• Relevant Laboratory Investigations Raised a. PTT could indicate: – Deficiency of factors: • • • – – – – – II V VIII IX X XI XII PK HMWK Fibrinogen Subtypes of v. WD (associated FVIII deficiency) Autoantibodies (inhibitors) against the above factors Anticoagulant therapy with heparin Anticoagulant drugs directly affecting thrombin (i. e. hirudin) Anticoagulant drugs (e. g. Warfarin) affecting Vit. K-dependant factor production (a. PTT is less affected than PT in this case as there are more non-vit. K-dependant factors involved in a. PTT clot formation) Vit. K-deficiency Liver disease DIC Lupus anticoagulants
Relevant Laboratory Investigations If a factor deficiency is suggested, further assays should be performed to determine which factor is deficient and its plasma concentration a. One-stage coagulation factor assays b. Two-stage coagulation factor assays
Laboratory Investigations Test Factor Deficiency v. WD Abnormally Increased PT Normal a. PTT Elevated only if FVIII is sufficiently reduced (Type 1, 2 N, 3) FVIII Corrected if FVIII is sufficiently reduced (Type 1, 2 N, 3) FIX Corrected - FXI Corrected - v. WF - Reduced Activity (Type 1, 2 A, 2 B, 2 M, 3) Mixing study with normal plasma
Factor Concentrates • Recombinant FVIII – Advate – Recombinate – Kogenate – Refacto • Recombinant FIX – Benefix • EHL FVIII – Eloctate • EHL FIX – Alprolix – Idelvion
Idelvion – Conjugated to bilirubin (hence green colouration) which extends half-life – More expensive than more recent products (£ 1. 40 per IU as opposed to £ 0. 83* per IU for Benefix) – Use must be cost-neutral * Note that due to competitive market forces, Pfizer reduced the cost of Benefix to £ 0. 27 per IU shortly after Idelvion was released on to the market
Future Extended Half-Life Clotting Factors • PEGylated clotting factors • Addition of Polyehtyleneglycol (PEG) to clotting factor • Polysialylated clotting Factors • Addition of polysialic acid to clotting factor Note: These two treatments essentially do the same thing using a different compound
Emicizumab/Hemlibra (ROCHE: ACE-910) Monoclonal Ab for treatment of Haemophilia A with, or without, inhibitors – Phase III clinical trial involved a serious adverse reaction – a patient died – Clinical trial was paused, and has now been reinstated – Should be on the market in the UK soon.
RNAi Technology • Fitusiran (Alnylam)
But the future is really…
Gene Therapy The first study to demonstrate that gene therapy works in humans: NEJM Nov 2011 AAV 8 vector containing FIX gene
Haemophilia A – Large gene, thus difficulties determining the ideal vector – Progress is steady, but lagging behind that for haemophilia B Haemophilia B – Smaller gene than haemophilia A, therefore easier to find a suitable vector – AAV vector (common cold) seem to be making the best progress, with several serotypes under study Many biotech companies engaged in this research, for example: • • Dimension Therapeutics Spark Therapeutics Bio. Marin Etc…
What could be more effective than Gene Therapy? In-utero Gene Therapy? – Are there ethical considerations in permanently altering the genetics of an unborn foetus? Germline Gene Therapy? – Are there ethical considerations in permanently altering the germline?
Why the need for new treatments? Contaminated Blood Disaster For the full speech click here