HEMOSTASIS AND THROMBOSIS Normal hemostasis results from wellregulated
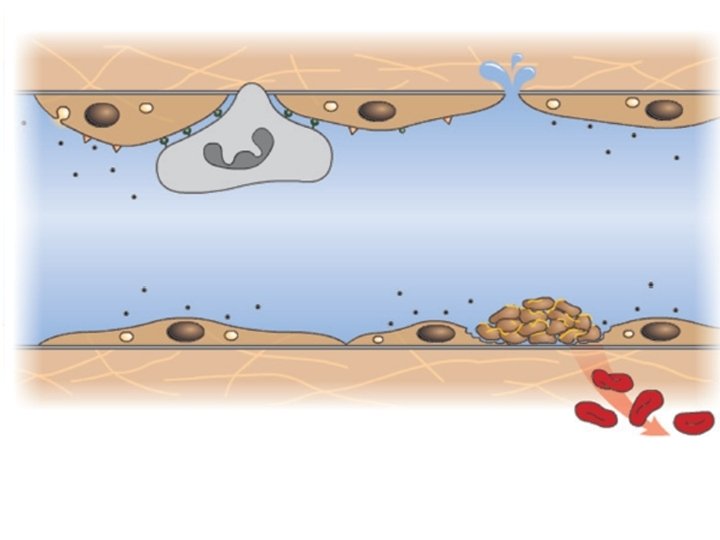

HEMOSTASIS AND THROMBOSIS Normal hemostasis results from well-regulated processes that maintain blood in a fluid, clot-free state in normal vessels, while inducing the rapid formation of a localized hemostatic plug at the site of vascular injury. The pathologic converse to hemostasis is thrombosis. Both hemostasis and thrombosis are dependent on three general components: -Vascular wall. -Platelets. -Coagulation cascade. Normal Hemostasis -After initial injury, there is a brief period of arteriolar vasoconstriction caused by endothelin (a potent endothelium-derived vasoconstrictor). -Injured endothelium exposed underlining highly thrombogenic subendothelial extracellular matrix (ECM), which allows platelets to adhere and activate, where platelets undergo a shape change and release secretory granules. These granules have recruited additional platelets (aggregation) to form a hemostatic plug; this is the process of (primary hemostasis).

Released tissue factor, a membrane-bound procoagulant factor synthesized by endothelium. It acts in conjunction with the secreted platelet factors to activate the coagulation cascade, culminating in activation of thrombin. In turn, thrombin cleaves circulating fibrinogen into insoluble fibrin, creating a fibrin meshwork deposition. Thrombin also induces further platelet recruitment to form secondary hemostasis. Endothelium exhibit antiplatelet, anticoagulant, and fibrinolytic properties; on the other hand, additionally they have procoagulant functions. Anti-thrombotic properties of (endothelial cells): 1 -Antiplatelet effects: An intact endothelium prevents platelets from meeting the highly thrombogenic subendothelial ECM. Nonactivated platelets do not adhere to the endothelium. If platelets are activated (e. g. , after focal endothelial injury), they are prevented from adhering to the surrounding uninjured endothelium by endothelial prostacyclin (PGI 2) and nitric oxide.

Normal hemostasis. A, After vascular injury, local neurohumoral factors induce a transient vasoconstriction. B, Platelets adhere (via Gp. Ib receptors) to exposed extracellular matrix (ECM) by binding to von Willebrand factor (v. WF) and are activated, undergoing a shape change and granule release. Released adenosine diphosphate (ADP) and thromboxane A 2 (TXA 2) lead to further platelet aggregation (via binding of fibrinogen to platelet Gp. IIb-IIIa receptors), to form the primary hemostatic plug.
 results")
C, Local activation of the coagulation cascade (involving tissue factor and platelet phospholipids) results in fibrin polymerization, "cementing" the platelets into a definitive secondary hemostatic plug that is larger and more stable than the primary plug and contains entrapped red cells and leukocytes. D, Counterregulatory mechanisms, such as release of t-PA (tissue plasminogen activator, a fibrinolytic product) and thrombomodulin (interfering with the coagulation cascade), limit the hemostatic process to the site of injury.

2 -Anticoagulant properties: These are mediated by membrane-associated, heparin-like molecules and thrombomodulin, a specific thrombin receptor. 3 -Fibrinolytic properties: Endothelial cells synthesize t-PA, promoting fibrinolytic activity to clear fibrin deposits from endothelial surfaces. Prothrombotic properties of (endothelial cells): Endothelial cells are also induced by cytokines (e. g. , tumor necrosis factor [TNF] or interleukin 1 [IL-1]) or bacterial endotoxin to secrete tissue factor, which activates the extrinsic clotting pathway. By binding to activated coagulation factors IXa and Xa, endothelial cells further augment the catalytic activities of these proteins. Finally, endothelial cells also secrete inhibitors of plasminogen activator (PAIs), which depress fibrinolysis. The balance between endothelial anti- and prothrombotic activities determines whether thrombus formation, propagation, or dissolution occurs.

The series of platelet events: -Platelets adhere to the ECM at sites of endothelial injury and become activated. -Upon activation, platelets secrete granule products (e. g. , ADP) and synthesize TXA 2. -Platelets also expose phospholipid complexes important in the intrinsic coagulation pathway. -Injured or activated endothelial cells release tissue factor to activate the extrinsic coagulation cascade. -Released ADP stimulates formation of a primary hemostatic plug, which is eventually converted (via ADP, thrombin, and TXA 2) into a larger definitive secondary plug. -Fibrin deposition stabilizes and anchors the aggregated platelets. Each reaction in the pathway results from the assembly of a complex composed of an enzyme (activated coagulation factor), a substrate (proenzyme form of coagulation factor), and a cofactor (reaction accelerator). These components are assembled on a phospholipid complex and held together by calcium ions. Thus, clotting tends to remain localized to sites where such an assembly can occur, for example, on the surface of activated platelets.
 to")
Sequential conversion of factor X to factor Xa, followed by factor II (prothrombin) to factor IIa (thrombin). The initial reaction complex consists of an enzyme (factor IXa), a substrate (factor X), and a reaction accelerator (factor VIIIa), all assembled on a platelet phospholipid surface. Calcium ions hold the assembled components together and are essential for the reaction. Activated factor Xa becomes the enzyme part of the second adjacent complex in the coagulation cascade, converting the prothrombin substrate to IIa using factor Va as the reaction accelerator.

Clotting is also controlled by two natural anticoagulants: • Antithrombins (e. g. , antithrombin III) inhibit the activity of thrombin and other serine proteases-factors IXa, XIa, and XIIa. Antithrombin III is activated by binding to heparin-like molecules on endothelial cells; hence, the usefulness of administering heparin in clinical situations to minimize thrombosis. • Proteins C and S are two vitamin K-dependent proteins that inactivate the cofactors Va and VIIIa. The activation of protein C by thrombomodulin was described earlier. Thrombosis: The pathologic converse to hemostasis is thrombosis. Pathogenesis: Three primary influences predispose to thrombus formation, the so-called Virchow triad: (1) Endothelial injury. (2) Stasis or turbulence of blood flow. (3) Blood hypercoagulability.
.")
The fibrinolytic system, illustrating various plasminogen activators and inhibitors (see text).

A-Endothelial injury is the dominant influence and by itself can lead to thrombosis. It is particularly important in thrombus formation in the heart and arterial circulation, for example, within the cardiac chambers when there has been endocardial injury (e. g. , myocardial infarction or valvulitis), over ulcerated plaques in severely atherosclerotic arteries, or at sites of traumatic or inflammatory vascular injury. Endothelial dysfunction may occur by: 1 - The hemodynamic stresses of hypertension. 2 -Turbulent flow over scarred valves. 3 -Bacterial endotoxins. Even relatively subtle influences such as homocystinuria, hypercholesterolemia, radiation, or products absorbed from cigarette smoke may be sources of endothelial injury and dysregulation.

Virchow's triad in thrombosis. Integrity of endothelium is the most important factor. Injury to endothelial cells can also alter local blood flow and affect coagulability. Abnormal blood flow (stasis or turbulence), in turn, can cause endothelial injury. The factors may act independently or may combine to promote thrombus formation.
 Disrupt laminar flow: bring")
B-Alterations in normal blood flow: -Stasis and turbulence induce: (1) Disrupt laminar flow: bring platelets into contact with the endothelium. (2) Prevent dilution of activated clotting factors by fresh-flowing blood. (3) Retard the inflow of clotting factor inhibitors and permit the build-up of thrombi. (4) Promote endothelial cell activation. -Turbulence and stasis: 1 -Ulcerated atherosclerotic plaques not only expose subendothelial ECM but also generate local turbulence. 2 -Abnormal aortic and arterial dilations called aneurysms cause local stasis and are favored sites of thrombosis. 3 -Myocardial infarctions not only have associated endothelial injury but also have regions of noncontractile myocardium, adding an element of stasis in the formation of mural thrombi. 4 -Mitral valve stenosis (e. g. , after rheumatic heart disease) results in left atrial dilation. In conjunction with atrial fibrillation, a dilated atrium is a site of profound stasis and a prime location for development of thrombi.
 increase resistance to flow and cause small vessel")
5 -Hyperviscosity syndromes (such as polycythemia) increase resistance to flow and cause small vessel stasis. 6 -Deformed red cells in sickle cell anemia cause vascular occlusions, with the resultant stasis predisposing to thrombosis. C-Hypercoagulability: -Defined as any alteration of the coagulation pathways that predisposes to thrombosis. -Contributes less frequently to thrombotic states. -Divided into primary (genetic) and secondary (acquired) disorders. The inherited (genetic) causes: -Mutations in the factor V gene and prothrombin gene are the most common. The characteristic alteration is a mutant factor Va that cannot be inactivated by protein C; as a result, an important antithrombotic counter-regulatory pathway is lost.

-Acquired causes: 1 -Hypercoagulability may be related to increased hepatic synthesis of coagulation factors and reduced synthesis of antithrombin III. 2 -In disseminated cancers, release of procoagulant tumor products predisposes to thrombosis. 3 -The hypercoagulability seen with advancing age may be due to increasing platelet aggregation and reduced PGI 2 release by endothelium. 4 -Smoking and obesity promote hypercoagulability. Thrombi may develop anywhere in the cardiovascular system: within the cardiac chambers, on valve cusps, or in arteries, veins, or capillaries. They are of variable size and shape, depending on the site of origin and the circumstances leading to their development. -Arterial or cardiac thrombi usually begin at a site of endothelial injury (e. g. , atherosclerotic plaque) or turbulence (vessel bifurcation). -Venous thrombi characteristically occur in sites of stasis.
 Factor V mutations Prothrombin")
CONDITIONS ASSOCIATED WITH AN INCREASED RISK OF THROMBOSIS Primary (Genetic) Factor V mutations Prothrombin mutation Antithrombin III deficiency Protein C or S deficiency Secondary (Acquired) 1 -High risk for thrombosis Prolonged bed rest or immobilization Myocardial infarction Tissue damage (surgery, fracture, burns) Cancer Prosthetic cardiac valves Disseminated intravascular coagulation Lupus anticoagulant 2 -Low risk for thrombosis Atrial fibrillation Cardiomyopathy Nephrotic syndrome Hyperestrogenic states Oral contraceptive use Sickle cell anemia Smoking

An area of attachment to the underlying vessel or heart wall, frequently firmest at the point of origin, is characteristic of all thrombi. -Arterial thrombi tend to grow in a retrograde direction from the point of attachment. -Venous thrombi extend in the direction of blood flow, that is, toward the heart. The propagating tail may not be well attached and, particularly in veins, is prone to fragment, creating an embolus. Lines of Zahn When formed in the heart or aorta, thrombi may have grossly (and microscopically) apparent laminations called lines of Zahn; these are produced by pale layers of platelets and fibrin that alternate with darker layers containing more red cells. Lines of Zahn are significant only in that they imply thrombosis at a site of blood flow; in veins or in smaller arteries, the laminations are typically not as apparent. Thrombi formed in the sluggish venous flow usually resemble coagulated blood (much like blood clotted in a test tube). Nevertheless, careful evaluation generally reveals irregular, ill-defined laminations.

Lines of Zahn

Right atrial mural thrombus with lines of Zahn

Mural thrombi and aortic thrombus When arterial thrombi arise in heart chambers or in the aortic lumen, they are usually applied to the wall of the underlying structure and are termed mural thrombi. Abnormal myocardial contraction (arrhythmias, dilated cardiomyopathy, or myocardial infarction) or injury to the endomyocardial surface (myocarditis, catheter trauma) leads to the formation of cardiac mural thrombi, while ulcerated atherosclerotic plaques and aneurysmal dilation are the precursors of aortic thrombus formation. Arterial thrombi Usually occlusive; the most common sites, in descending order, are coronary, cerebral, and femoral arteries. It caused by atherosclerotic plaque, vascular injury (vasculitis, trauma) The thrombi are firmly adherent to the injured arterial wall and are gray-white and friable, composed of a tangled mesh of platelets, fibrin, erythrocytes, and degenerating leukocytes.

Low power view of thrombosed coronary artery. Note previous narrowing of the lumen by atherosclerosis. Most such events arise from rupture or fissuring of a vulnerable placque.

Early organization of coronary thrombus. Subintimal tissue is at bottom. Note in growth of fibroblasts and capillaries into the thrombus
 Is almost invariably occlusive; and often creates a long cast of")
Venous thrombosis (phlebothrombosis) Is almost invariably occlusive; and often creates a long cast of the vein lumen. Because these thrombi form in the slowly moving venous blood, they contain more enmeshed erythrocytes and are therefore known as (red, or stasis), thrombi. Phlebothrombosis most commonly (90% of cases) affects the veins of the lower extremities. Less commonly, venous thrombi may develop in the upper extremities, periprostatic plexus, or ovarian and periuterine veins. At autopsy, postmortem clots may be mistaken for venous thrombi, but the Postmortem clots are gelatinous with a dark red dependent portion where red cells have settled by gravity, and a yellow "chicken fat" supernatant; they are usually not attached to the underlying wall. In contrast, red thrombi are firmer, almost always have a point of attachment, and on transection reveal vague strands of pale gray fibrin. Vegetations (infective endocarditis) Thrombi may form on heart valves. Bacterial or fungal blood-borne infections may lead to valve damage and the development of large thrombotic masses.

Fate of the Thrombus: If a patient survives, thrombi undergo some combination of the following four events in the ensuing days or weeks: 1 -Propagation: The thrombus may accumulate more platelets and fibrin (propagate), eventually obstructing some critical vessel. 2 -Embolization: Thrombi may dislodge and be transported to other sites in the vasculature. 3 -Dissolution: Thrombi may be removed by fibrinolytic activity such as (t-PA), only for a short time after thrombi form. 4 -Organization and recanalization: Thrombi may induce inflammation and fibrosis (organization) and may eventually become recanalized (re-establish vascular flow), or they may be incorporated into a thickened vascular wall. Such recanalization can potentially convert the thrombus into a vascularized mass of connective tissue that is eventually incorporated as a subendothelial swelling into the vessel wall.
 occlusion")
Resolution Propagation – (complete) occlusion

Fragmentation and Embolization

Organization – Fibroblast proliferation Organization – Endothelial cell differentiation

Organization and re-canalized thrombus End-stage of organization of a thrombus. The thrombus (left two-thirds of field) is replaced by connective tissue with many newly formed channels. If these channels reestablish (arrow) the continuity of the original vessel, blood may flow at a rate adequate to maintain perfusion. Note calcification (black crystals (circle).
 they cause")
Clinical Correlations: Venous Thrombosis versus Arterial Thrombosis. Thrombi are significant because: (1) they cause obstruction of arteries and veins. (2) they are possible sources of emboli. A far graver consequence is that they may embolize to the lungs, causing death. Congestion and edema usually occurred. Conversely, arterial thrombi can embolize the critical sites, such as coronary or cerebral vessels. -Venous thrombosis (phlebothrombosis): -Superficial venous thrombi of the leg usually occur in the saphenous system, particularly when there are varicosities. Such thrombi may cause local congestion and cause swelling, pain, but they rarely embolize. -Deep thrombi in the larger leg veins at or above the knee joint (e. g. , popliteal, femoral, and iliac veins) are more serious because they may embolize. They may cause local pain and edema.

Marked passive congestion of the left lower leg in a patient with a deep venous thrombus Surgical dissection of the thrombus. Cardiac and arterial thrombosis In addition to the obstructive consequences, cardiac and aortic mural thrombi can also embolize peripherally. Virtually any tissue may be affected, but the brain, kidneys, and spleen are prime targets because of their large flow volume.

Aorta Aneurysm with thrombus Kidney Atherosclerotic Abdominal aortic Aneurysm

Mural thrombi. A, Thrombus in the left and right ventricular apices, overlying white fibrous scar.

Low-power view of an artery with an old thrombus. A, H&E-stained section. B, Stain for elastic tissue. The original lumen is delineated by the internal elastic lamina (arrows) and is totally filled with organized thrombus, now punctuated by a number of recanalized channels (white spaces).

Organized venous thrombus en um d. L we rro Na

Organized venous thrombus Narrowed Lumen Muscular layer

Organized venous thrombus Proliferative fibroblast Smooth muscle cells Muscular layer Narrowed Lumen Organized thrombus

Saddle thrombus of the pulmonary trunk often leads to sudden death.
")
Pulmonary thrombus – organised (groossly)

Fat emboli stained red

EMBOLISM Embolus is a detached intravascular solid, liquid, or gaseous mass that is carried by the blood to a site distant from its point of origin. 99% of all emboli represent some part of a dislodged thrombus (thromboembolism). Rare forms of emboli include droplets of fat, bubbles of air or nitrogen, atherosclerotic debris (cholesterol emboli), tumor fragments, bits of bone marrow, or foreign bodies such as bullets. 1 -Pulmonary Thromboembolism In more than 95% of instances, venous emboli originate from deep leg vein thrombi above the level of the knee. They are carried through larger channels and usually pass through the right side of the heart into the pulmonary vasculature. Depending on the size of the embolus, it may occlude the main pulmonary artery, impact across the bifurcation (saddle embolus), or pass out into the smaller, branching arterioles. -Most pulmonary emboli (60% to 80%) are clinically silent because they are small. -Sudden death: right ventricular failure (cor pulmonale), or cardiovascular collapse occur when 60% or more of the pulmonary circulation is obstructed with emboli.

Embolus derived from a lower extremity deep venous thrombosis and now impacted in a pulmonary artery bifurcation (saddle embolus), .
 arise")
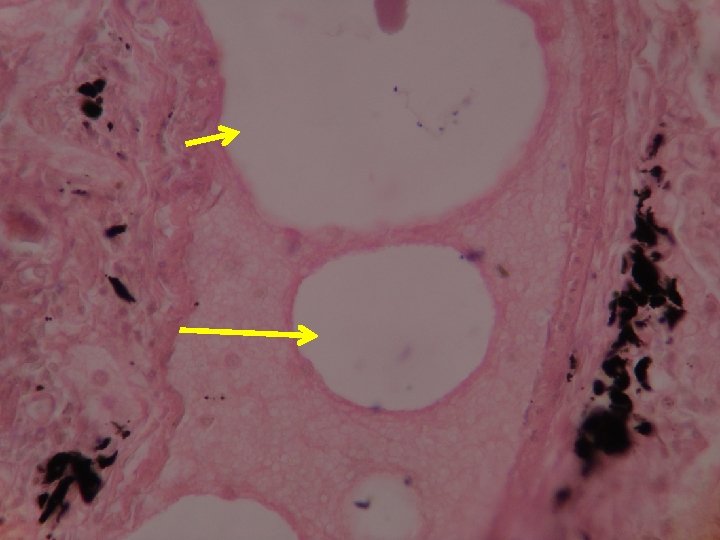
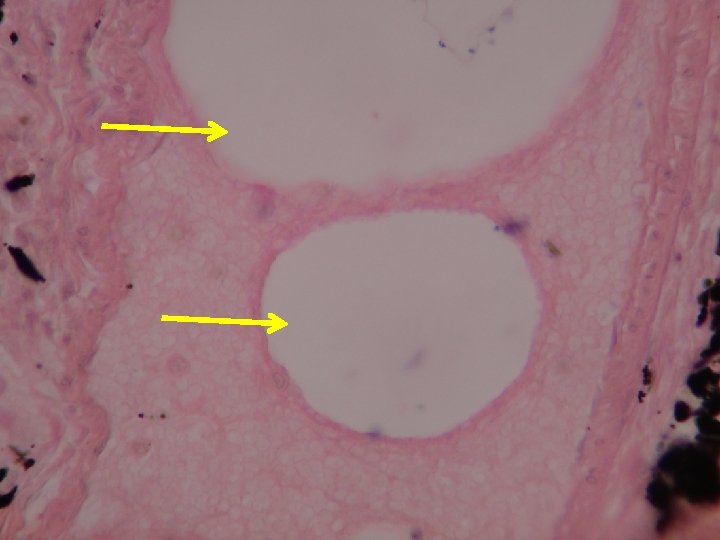
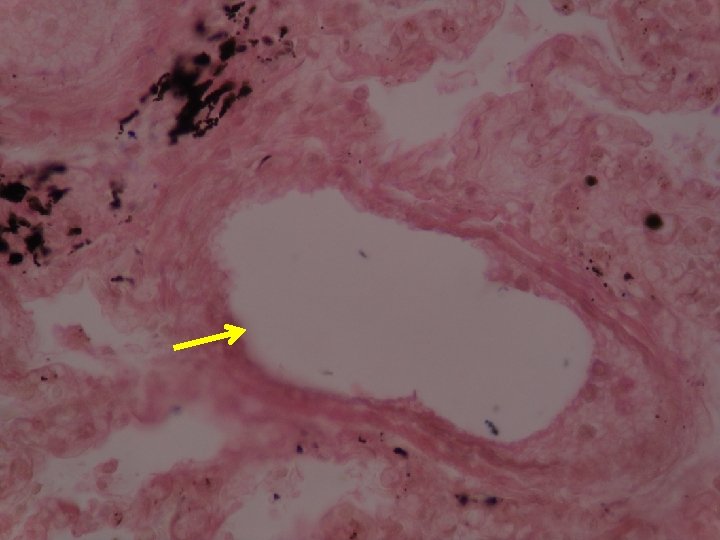
2 -Systemic thromboembolism Refers to emboli traveling within the arterial circulation. Most (80%) arise from Intracardiac mural thrombi: -Left ventricular wall infarcts. -Dilated left atria (e. g. , secondary to mitral valve disease). The remainder largely originate from thrombi associated with: -Ulcerated atherosclerotic plaques. -Aortic aneurysms. Sites of embolization -Venous emboli tend to lodge primarily in one vascular bed (the lung). -Arterial emboli can travel to a wide variety sites of the body, lower extremities (75%) and the brain (10%), with the intestines, kidneys, and spleen involved to a lesser extent. Fat Embolism Microscopic fat globules may be found in the circulation after fractures of long bones (which have fatty marrows) or, rarely, in the setting of soft tissue trauma and burns. Presumably, the fat is released by marrow or adipose tissue injury and enters the circulation by rupture of the marrow vascular sinusoids or rupture of venules.

H&E stain Fat embolism Special stain

Fat embolism after fracture of the leg
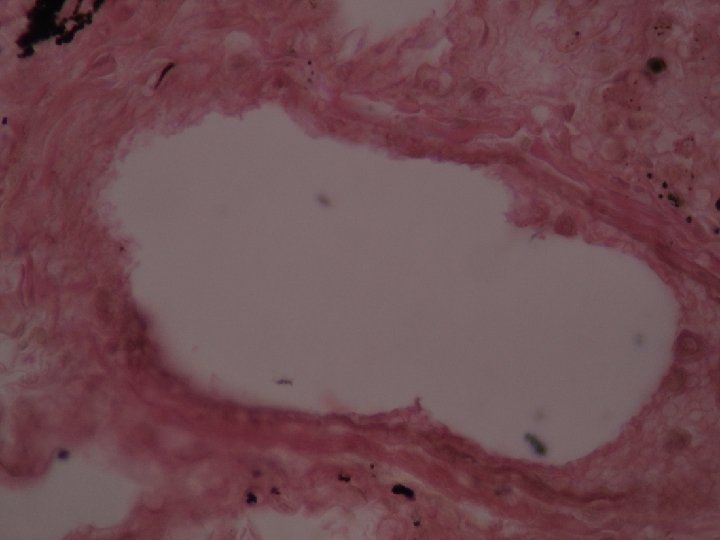

Marrow emboli

Hematopoietic tissue Bone marrow embolus in the pulmonary circulation. The cellular elements on the left side of the embolus are hematopoietic precursors, while the cleared vacuoles represent marrow fat. The relatively uniform red area on the right of the embolus is an early organizing thrombus.
 -Infusion of amniotic fluid (and its contents) into the maternal")
Amniotic fluid emboli (squames) -Infusion of amniotic fluid (and its contents) into the maternal circulation via a tear in the placental membranes and rupture of uterine veins. -Microscopically: presence in the pulmonary microcirculation of squamous cells shed from fetal skin. and mucin derived from the fetal respiratory or gastrointestinal tracts. There is also marked pulmonary edema.

Tumor embolus
 and surround themselves with")
Tumor embolism: In circulation, tumor cells aggregate in clumps (1) and surround themselves with a coat of fibrin (2), forming a tumor embolus that lodges in pre-capillary arterioles.

Pulmonary microembolism
 Recurrent embolism: Pulmonary embolisms become organized into")
Strand of organized embolism, (cross section view) Recurrent embolism: Pulmonary embolisms become organized into stranded structures. These strands can trap smaller emboli, causing occlusion of a branch of the pulmonary artery. In this manner, a peripheral embolism may become a central embolism.

INFARCTION An infarct is an area of ischemic necrosis caused by occlusion of either the arterial supply or the venous drainage in a particular tissue. -Red infarcts occur (1) with venous occlusions. (2) in tissues with dual circulations such as lung and small intestine. (3) -White infarcts occur with arterial occlusions, or in solid organs (such as heart, spleen, and kidney). (single circulation). Causes: -Mostly the infarcts result from thrombotic or embolic events, and almost all result from arterial occlusion. -Local vasospasm. -Swelling of an atheroma. -Compression of a vessel by tumor. -Twisting of the vessels (e. g. , in testicular torsion or bowel volvulus), -Compression of the blood supply by edema. -Entrapment in a hernia sac.

In spongy organs, by comparison, the hemorrhage is too extensive to permit the lesion ever to become pale. Over the course of a few days, however, it does become firmer and browner, reflecting the development of hemosiderin pigment. Red infarcts. A, Hemorrhagic, roughly wedge-shaped pulmonary infarct (red infarct).
 infarcts of small bowels")
Hemorrhagic (red) infarcts of small bowels

Hemorrhagic infarcts of the lung

Coagulative necrosis, kidney infarction This is the typical pattern with ischemia and infarction (loss of blood supply and resultant tissue anoxia). Here, there is a wedge-shaped pale area of coagulative necrosis (infarction) in the renal cortex of the kidney.

Infarctions in the spleen Infarction of many internal organs leads to a "pale" infarct with a wedgeshaped gross appearance (conical in 3 dimensions) from occlusion of a branching blood supply. Here are splenic infarcts in a patient with infective endocarditis. Portions of the vegetations have embolized to the spleen. These infarcts are typical of ischemic infarcts: they are based on the capsule, pale, and wedge-shaped.
.")
White infarcts. B, Sharply demarcated pale infarct in the spleen (white infarct).
- Slides: 62