Spi DME meeting Nijmegen May 2007 Stefano Sanvito

, April ‘ 07 Mr. Sankar Kesanakurthi (U. Hiderabad),")





)")
")



 Molecule Green")
 Ø")














ETHYLENEDIAMINO-TM Where TM could be :")
 Big E (e. V) Comparison between the DOS of the")
")
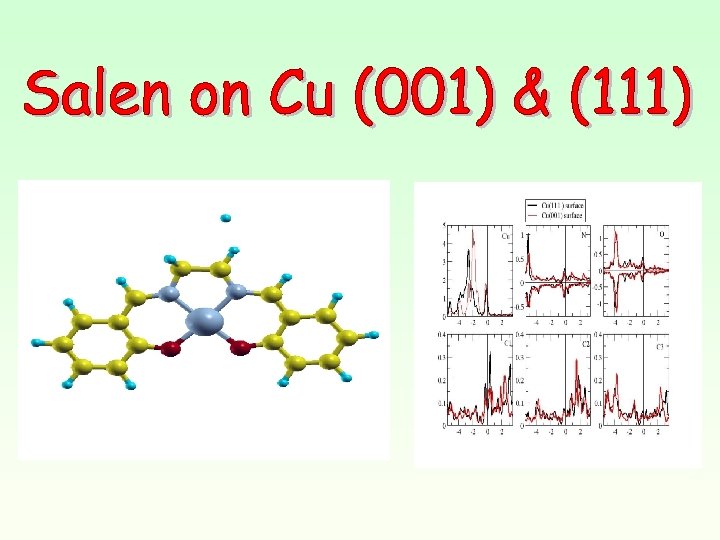
 Cu-salen on Cu(111) DOS (arb. units) Molecule on Cu surfaces (un-relaxed)")
 surface Unrelaxed structure Relaxed structure")
 Cu E (e. V) 4 s 13")
 Zn E (e. V) 4 s 23 d 10")
 Co E (e. V) 4 s 23 d 7")
 Ni 4 s 23 d 8 E (e. V)")


 (b) Zn (c) (d) EF-0. 2 e. V < EF EF <")


- Slides: 45
Spi. DME meeting, Nijmegen, May 2007 Stefano Sanvito and Nadjib Baadji Computational Spintronics Group School of Physics and CRANN, Trinity College
People Dr. Nadjib Baadji (Uni. Strasbourg), April ‘ 07 Mr. Sankar Kesanakurthi (U. Hiderabad), April ‘ 07 Visits Sanvito to Hamburg (Feb. 2007)
u A simple model for transport u Ab initio transport theory u SP-STM for molecules u Salen on Cu u Outlook
m. L V 0 GL e GR m. R
m. L e GR V m. R GL V≠ 0 In equilibrium Out of equilibrium 2
m. L e GR V m. R GL V≠ 0 2|EF- |
e = e 0 + U (N - f (e - m 0 )) L L R R m. L e GR V m. R GL V≠ 0
E +e. V/2 F E -e. V/2 F E T(E)
I
H= HM+H 0 +…. H 1 HLM HRM H 1 R L H 0 H 0 H 0 HM (n) HM+SL (E)+SR(E)
Lead’s Self-energy A. R. Rocha and S. Sanvito, PRB 70, 094406 (2004) Molecule Green function Density Matrix Current
KS-DFT Hamiltonian We implemented NEGF in Siesta Ø Localized multiple-z Pseudo-atomic orbitals (non-orthogonal) Ø Optimized Pseudopotential Ø Super-cells with up to 2, 000 atoms D. Sánchez-Portal, P. Ordejón, E. Artacho, and J. M. Soler, Int. J. Quant. Chem. 65, 453 (1997)
http: //www. smeagol. tcd. ie/ Mailing list http: //lists. tchpc. tcd. ie/listinfo/smeagol-discuss A. R. Rocha et al. , Phys. Rev. B 73, 085414 (2006); Nature Materials 4, 335 (2005)
Problems with molecular transport Ni point contacts C. Toher et al. , PRL 95, 146402 (2005) A. R. Rocha et al. , cond-mat/0701512 Fe/Mg. O TMR junction I. Rungger et al Spin Torque M. Stamenova et al. , in preparation DNA transport Molecular Spin valves A. R. Rocha et al. , in preparation Nature Mat. 4, 335 (2005)
80 n. A Au on Au 40 n. A V=250 m. V d=0. 4 nm 0 n. A 100 n. A Ni on Ni 50 n. A V=250 m. V d=0. 4 nm 0 n. A
20% 10% -10% I to tip 0% -30% -40% -30% I I P= I +I -55% -45% I from tip -70% -60% 500 m. V -250 m. V -500 m. V
Does the GMR mirror the polarization ? 10% -10% P= I I I +I -30% 250 m. V -10% -15% -20% I P I AP R= I AP
20% 500 m. V 10% 250 m. V -45% -250 m. V -50% -500 m. V
V=0
TIP M+S V=0
S S tip V
TIP M+S V=0
TIP M+S V=400 m. V Current to the tip
TIP M+S V=0
TIP M+S V=-400 m. V Current to the S+M
ØDirect calculations of the tunneling currents are possible and include: ü Electronic Structure of the tip ü Tip to sample interaction ü Charging of the moleculae ü Accurate determination of the spin-polarization ü Non-collinear spin ü Spin-orbit Ø Some prospects of investigating the bonding of molecules on magnetic surfaces
Molecule C 2 C 3 C 1 N, N'-BIS(SALICYLIDENE)ETHYLENEDIAMINO-TM Where TM could be : Cu, Zn, Ni or Co
Small DOS (arb. units) Big E (e. V) Comparison between the DOS of the Salen molecule and the hypothetical small molecule
Big Small E (e. V)
Cu-salen on Cu(001) Cu-salen on Cu(111) DOS (arb. units) Molecule on Cu surfaces (un-relaxed) E (e. V)
Relaxation on Cu(001) surface Unrelaxed structure Relaxed structure
DOS for different TM-salen DOS (arb. units) Cu E (e. V) 4 s 13 d 10
DOS (arb. units) Zn E (e. V) 4 s 23 d 10
DOS (arb. units) Co E (e. V) 4 s 23 d 7
DOS (arb. units) Ni 4 s 23 d 8 E (e. V)
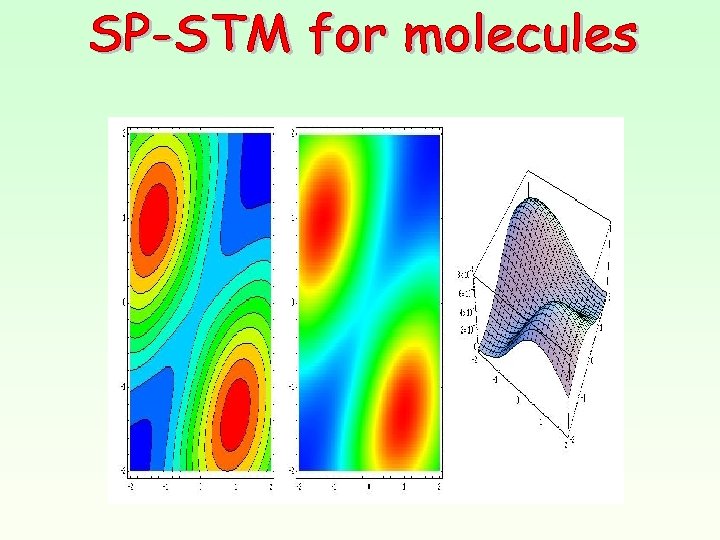
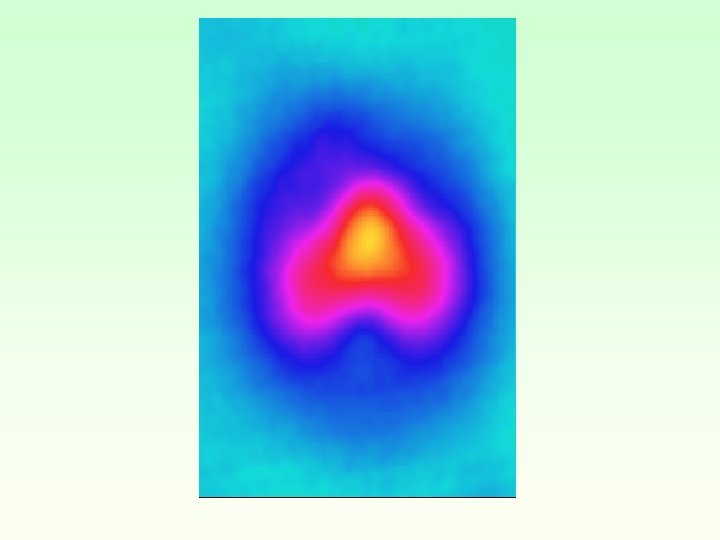
Simulation STM images Free Cu-Salen EF-0. 2 e. V < EF EF < EF +0. 2 e. V I molecule to tip I tip to molecule
Constant current STM images Cu-Salen un-relaxed EF-0. 2 e. V < EF EF < EF +0. 2 e. V I molecule to tip I tip to molecule
Cu (a) (b) Zn (c) (d) EF-0. 2 e. V < EF EF < EF +0. 2 e. V I molecule to tip I tip to molecule
ØThis is very much work in progress ü First find the right atomic configuration ü Then simulate the current ü Compare the images for different TM ü Hopefully they will compare with experiments
integral of the DOS near Ef (pos. & neg. bias Cu DOS in free mole. and in mole. on Cu (001) L-resolved DOS for Cu atom in L-resolved DOS for Zn atom in t