Primary lipid disorders A Amouzegar MD Research Institute


 particle")



 Total cholseterol: 600 -1000,")

 Drug therapy HMG-COA reductase inhibitor - Bile Acid sequestrants-Niacin •")






")




 •")

 - AD co •")



- AR • Moderate to severe")



")
 Dietary therapy Estrogen therapy Drug")



 het(1 in 500) • LPL deficiency")






 Causes a chylomicronemia")




 & LDL N & HDL L • TG-rich VLDL")








- Slides: 66
Primary lipid disorders A. Amouzegar. MD Research Institute For Endocrine Sciences Endocrine Research Center
AGENDA • • • Familial hypercholesterolemia Familial defective Apo B 100 Polygenic hypercholesterolemia Familial combined hyperlipidemia Familial dysbetalipoproteinemia Lipoprotein Lipase deficiency Apo Lipoprotein Cll deficiency Familial hyper triglyceridemia Familial Hypoalphalipoproteinemia
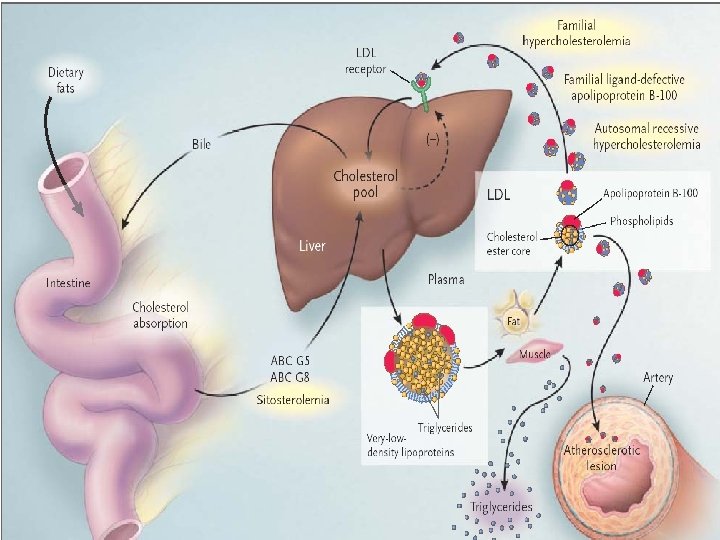
General structure of lipoproteins: schematic representation of a very-low-density lipoprotein (VLDL) particle
Small VLDL particles, IDL particles, and LDL particles may be taken up by peripheral tissues to deliver nutrients, cholesterol, and fat-soluble vitamins Free fatty acids liberated by the action of LPL are available to adipose tissue for storage and to other tissues (e. g. , skeletal muscle, heart) for use as energy substrates
Familial hypercholesterolemia • Caused by mutations in the LDL receptor gene that result in LDL receptor malfunction or absence in cells of the liver and peripheral tissues, leading to elevation of plasma LDL and TC concentrations • Plasma cholesterol concentrations are typically elevated 2 fold to 3 fold above average in heterozygous subjects and 3 fold to 6 fold in homozygous subjects • Heterozygosits: cholesterol>300 mg/dl LDL-C >250 mg/dl, , TG are not elevated-(1 in 5 oo) • Hyperlipidemia is present at birth
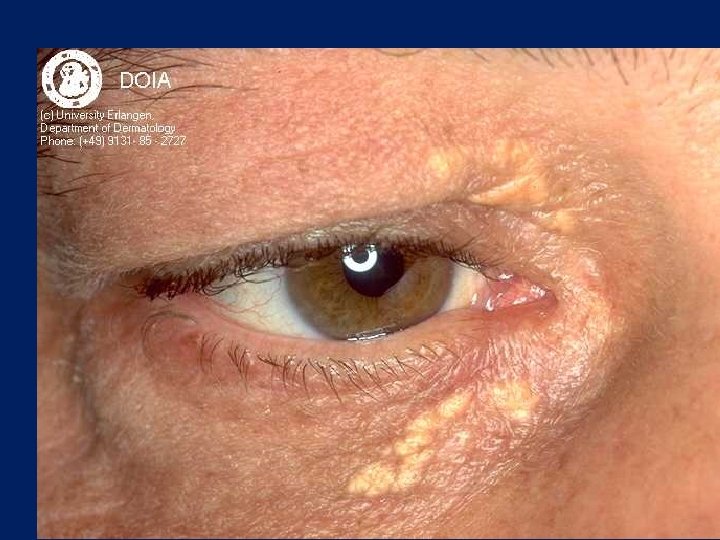
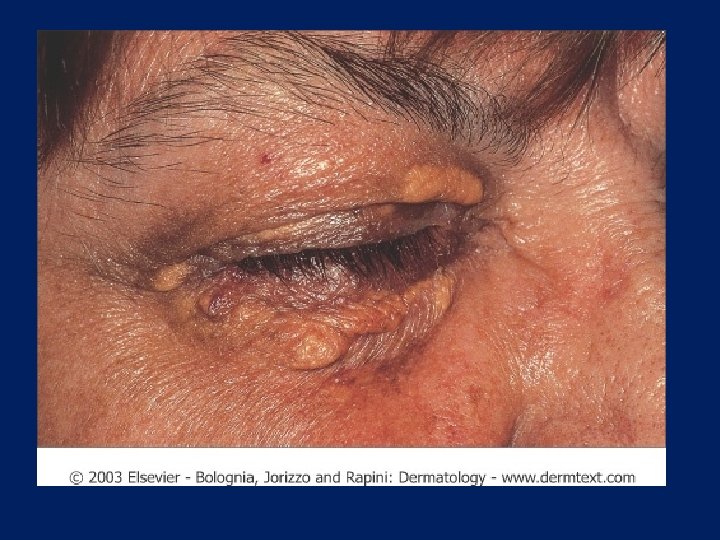
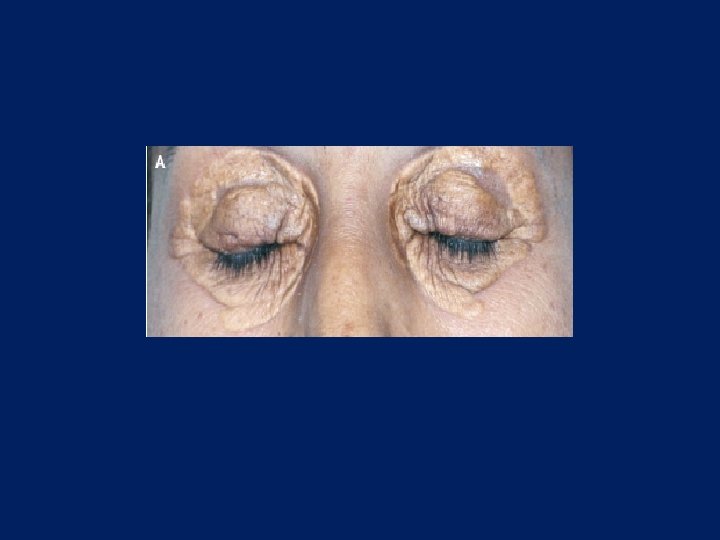
Characteristic physical finding: • Tendon xanthoma usually on the Achilles tendons or extensor tendons of the hands • Xanthelasma • premature arcus cornea • Many affected subjects have no physical findings • Premature CAD is common • The age of onset of coronary disease is about 45 years in men and women who are heterozygous for FH
Cont’ • Homozygote : is a rare (1 in 106) Total cholseterol: 600 -1000, LDL-C: 550 -950 • These subjects come to clinical attention early in life because of the appearance of xanthomas younger than the age of 10, marked hypercholesterolemia apparent at birth, or premature CHD
Pathogenesis and Diagnosis • • • Autosomal dominant Mutations in LDL receptor gene Diagnosis : The diagnosis of FH is primarily a clinical diagnosis because tests to detect one of the many LDL receptor gene mutations or to demonstrate diminished LDL receptor function are performed only in specialized research laboratories High plasma levels of total chol and LDL normal TG, tendon xanthoma and familial history of premature CHD
Treatment Diet modification(5 -15%) Drug therapy HMG-COA reductase inhibitor - Bile Acid sequestrants-Niacin • LDL apheresis • • •
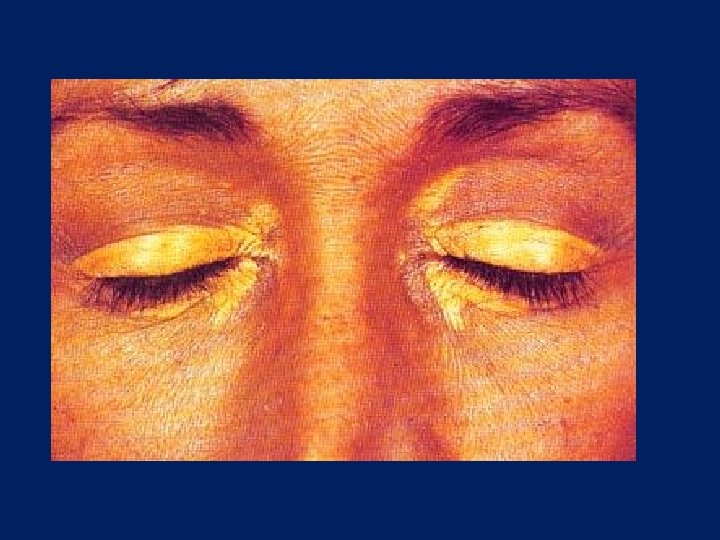
Xanthelasma
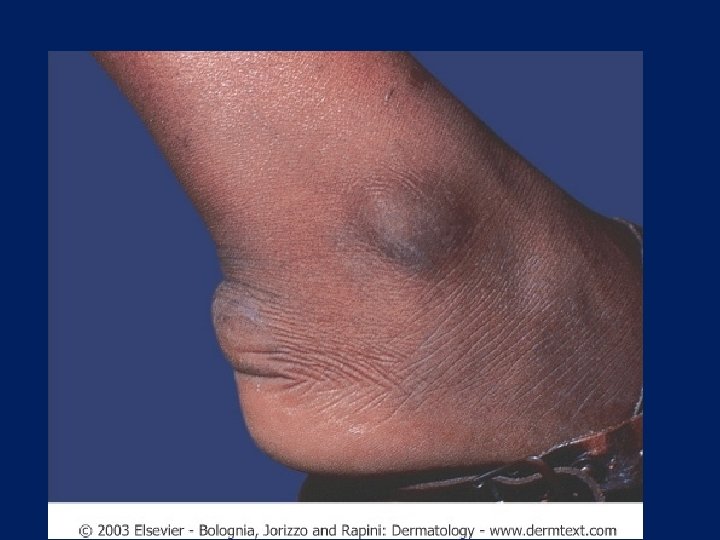
Achilles Tendon Xanthoma
Tendon Xanthoma
Metacarpophalangial Tendon Xanthoma
Tendon Xanthoma
Tuberous xanthomas
Xanthoma palpebrarum(Tuberous xanthomas)
premature arcus cornea
Familial defective Apolipoprotein. B 100 • AD • Frequency (1 in 500 to 1 in 750) • The clinical features of heterozygous familial defective apo-B 100 overlap extensively with those of heterozygous FH and include isolated elevations of plasma LDL, tendon xanthomas, xanthelasmas, and premature CHD
pathogenesis • Familial defective apo-B 100 is a relatively common disorder caused by a mutation in apo-B 100, the ligand that binds LDL to the LDL receptor • It results in high plasma LDL and total cholesterol levels • The less severe phenotype is related to the fact that the binding of apo-B to LDL receptors is defective but not totally whereas the apo-E–mediated clearance of remnant particles, which is impaired in FH, is normal in persons with familial defective apo-B 100
diagnosis • The diagnosis of familial defective apo-B 100 is suggested by the presence of increased plasma LDL-C and normal triglyceride levels, especially in a patient with tendon xanthomas and a family history of premature CHD • Without specialized testing, however, familial defective apo-B 100 is clinically indistinguishable from FH
Treatment • Similar to that heterozygots FH • Diet modification( low fat-low cholesterol) • Drug therapy • HMG-COA reductase inhibitor
Polygenic Hypercholesterolemia • In another 5% of patients, the cause of the hypercholesterolemia appears to involve combinations of multiple genetic and environmental factors • Other genetic factors that contribute to hypercholesterolemia can involve physiologic processes that influence cholesterol absorption, bile acid metabolism, or intracellular cholesterol metabolism • Polygenic hypercholesterolemia is diagnosed by excluding other primary genetic causes, by the absence of tendon xanthomas, and by demonstrating that hypercholesterolemia is present in no more than 10% of first-degree relatives
Familial combined Hyperlipidemia • A commo n disorder (0. 5%-2%) - AD co • A common disorder of unknown genetic cause associated with elevations of plasma cholesterol and triglyceride levels and increased susceptibility to CHD • The most common dyslipidemia syndrome in families of survivors of premature MI • Association with metabolic syndrome • Neither its genetic cause nor its metabolic pathogenesis is clear
diagnosis • The features include moderate elevations of plasma cholesterol or triglycerides, or both, within subjects of an affected kindred • The predominant lipid abnormality (hypertriglyceridemia, hypercholesterolemia, or both) can vary among affected family members or in a single person over time
Cont • Variability in the type of dyslipidemia is a useful hint that the subject might have this disorder • Levels of HDL-C are often moderately decreased, especially in the setting of increased plasma triglycerides • Xanthomas are not a feature of familial combined hyperlipidemia
Treatment • Weight reduction • Diet modification • Drug therapy: directed at the predominant lipid abnormality
Familial dysbetalipoproteinemia • An uncommon disorder(1 in 10. 000)- AR • Moderate to severe hypertriglyceridemia and hypercholesterolemia (300 -400 mg) • LDL is always reduced- HDL is normal • Premature vascular disease and CAD • Requires a secondary exacerbating metabolic factor (either genetic or environmental) for expression of the phenotype
Clinical Features • More common in men • Not manifested in women until after menopause • Tendon xanthomas and xanthelasma occur in some • Tuberus or tubero eruptive xanthoma • Palmar xanthoma : pathogonomic • Coexisting metabolic conditions that exacerbate such as obesity, alcohol consumption, diabetes mellitus, and hypothyroidism, are often present
pathogenesis • Caused by mutant apo. E • The clearance defect is caused by mutant apo. E that binds defectively to remnant receptors, including LDL receptors • Electrophoresis(broad band B - lipoprotein) • Plasma accumulation of cholesterol rich remnants of VLDL, IDL and chylomicron particles
Diagnosis • Should be suspected in patients with moderately severe, nearly equal (based on values expressed as mg/d. L) elevations in both plasma triglyceride and cholesterol concentrations • Typically the cholesterol and triglyceride levels are in the range of 300 -400 mg/d. L • There is often no family history of hyperlipidemia or premature CHD • The presence of palmar or tuberous xanthomas makes the diagnosis highly likely
• The directly measured VLDL cholesterol-toplasma triglyceride ratio (lipid values in mg/d. L) is a useful screen; this ratio is usually greater than 0. 3 The normal ratio is typically about 0. 2
Treatment • • • Treatment of coexisting condition (obesity-Dm-hypothyroidism-alcohol) Dietary therapy Estrogen therapy Drug therapy(niacin-fibric acid)
• Palmar xanthoma
Tuberous Xanthoma
Tuberous xanthomas
Lipoprotein lipase deficiency • AR disorder-hom(1 in 106) het(1 in 500) • LPL deficiency is usually recognized in infancy or childhood as a chylomicronemia syndrome • Severe hypertriglyceridemia • Heterozygot : mild to moderate TG • Recognized in infancy or childhood • Recurrent abdominal pain and Pancreatitis • Pain syndrome(TG>2000 -Eruptive xanthoma -Lipemia retinalis • The plasma may be visibly lipemic; plasma that has been refrigerated overnight might have a cream-like layer on the top, representing chylomicrons • If there is a turbid plasma infranatant, there is a high concentration of VLDL • Premature CHD is not prominent feature
pathogenesis and diagnosis • Mutations in the LPL gene • Plasma TG usually >1000 mg/dl • Eruptive xanthoma , lipemia retinalis and pancreatitis : TG level>2000 mg/dl • Accumulation of triglycerides in tissue reticuloendothelial cells can lead to hepatomegaly and splenomegaly • Established by absence of lipase activity after heparin administration • Heparin, when infused intravenously, displaces LPL from its binding sites on HSPG in capillary endothelium and releases it into plasma, which can be assayed for lipase activity
Treatment • Initial stage of treatment : fat free diet • Goal of therapy : TG<1000 mg/dl • Secondary causes should be screened and treated • Drug therapy : largely ineffective • however, clofibrate, gemfibrozil, fenofibrate, or niacin can lower VLDL production and lessen the severity of the hypertriglyceridemia • Orlistat lowers triglyceride levels significantly in some patients with severe hypertriglyceridemia
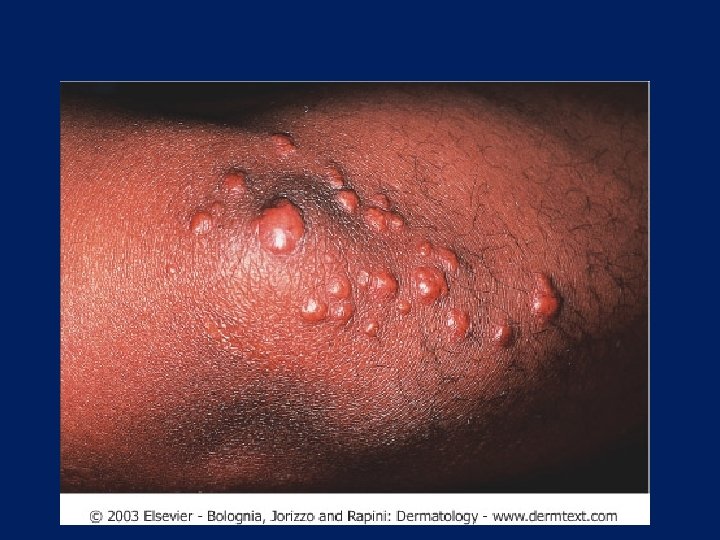
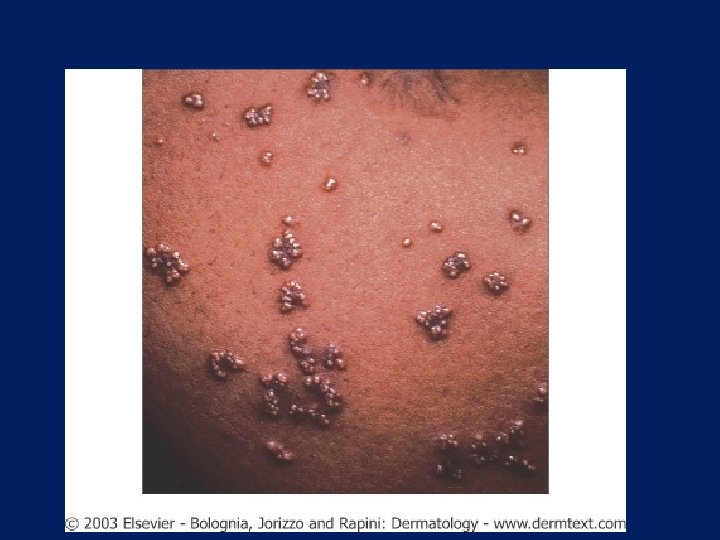
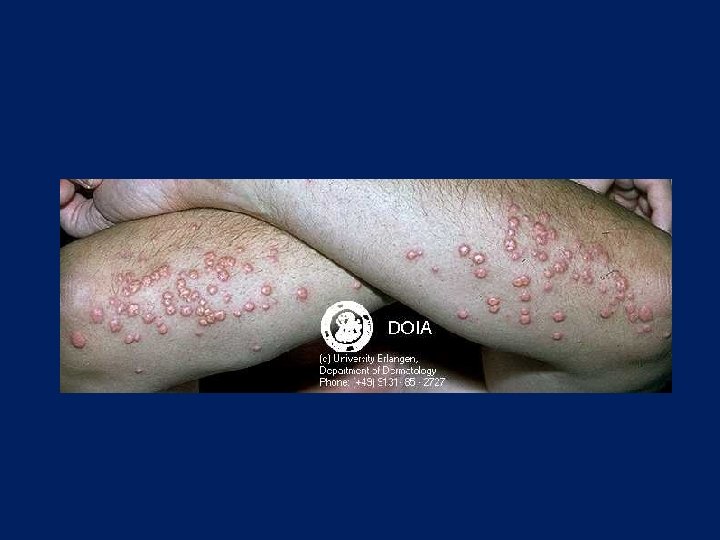
Eruptive Xanthoma
Eruptive Xanthoma
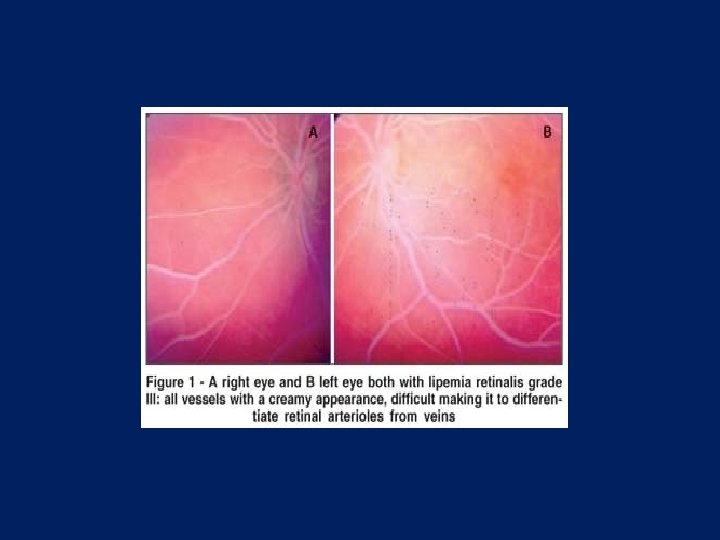
Lipemia retinalis
Lipemic plasma
Apo lipoprotein CII deficiency disorder • • AR disorder(1 in 106) Causes a chylomicronemia syndrome similar to that in LPL deficiency Plasma triglyceride levels are usually severely elevated ( 1000 mg/d. L), reflecting the accumulation of chylomicrons, VLDL, or both lipoproteins in the plasma This hyperlipoproteinemia results from the lack of apo-CII, an activating cofactor for LPL, which causes a functional LPL deficiency
The features: • • • As in LPL deficiency, the features include pancreatitis or recurrent bouts of abdominal pain in children or young adults and, after a 12 -hour fast, lipemic serum with chylomicrons that rise to the surface of serum that has stood undisturbed for 10 to 12 hours TG usually>1000 mg/dl Pancreatitis
Diagnosis • Requires specialized testing to demonstrate that apo-CII is absent on electrophoresis of the plasma apolipoproteins or that the plasma is incapable of activating LPL in vitro
Treatment • Is identical to that of primary LPL deficiency with the exception that severe hypertriglyceridemia in apo-CII-deficient patients with pancreatitis can be treated with transfusions of plasma, which contains apo-CII
Familial hypertriglyceridemia • Familial hypertriglyceridemia is characterized by increased plasma concentrations of triglyceriderich VLDLs, which cause elevations of plasma triglycerides but not plasma cholesterol levels • Appears to be caused by overproduction of VLDL triglycerides in the presence of near-normal apo. B production, which leads to the secretion of large, triglyceride-rich
Clinical Features • TG(200 -500) & LDL N & HDL L • TG-rich VLDL • Elevated TG usually not evident until adulthood • Exacerbated by secondary factors (e. g. , insulin resistance) that lead to VLDL overproduction can exacerbate the syndrome • Xanthoma usually not present. • Obesity & insulin resistance are common
pathogenesis and diagnosis • Overproduction of VLDL triglycerides. • • Genetic defect : AD Can be diagnosed in half of first degree relatives. • Difficult to distinguished from familial combined hyperlipidemia. • Increased risk of CHD?
Diagnosis • should be suspected in patients with increased plasma triglyceride levels and normal plasma cholesterol levels • The disorder can be diagnosed only if hypertriglyceridemia is found in half of the firstdegree relatives at risk, and it can be difficult to distinguish from familial combined hyperlipidemia • Elevated VLDL levels impart a cloudy appearance to plasma after overnight refrigeration
Treatment • • Dietary fat restriction. Secondary disorders should be screened Drug may be useful (Niacin-Gemfibrozil)
Familial Hypoalphalipoproteinemia • The AD • Manifested by low plasma HDL-C levels, normal LDL-C and fasting triglyceride levels, and an increased risk of premature CHD • The diagnosis is suggested by HDL-C levels that are less than the 10 th percentile ( 30 mg/d. L) in men or women (40 mg/d. L)
physical findings • There are no characteristic physical findings, but there is often a family history of low HDL-C levels and premature CHD • The genetic and metabolic defects that lead to low plasma HDL levels are unknown, but it appears that up to 50% of low HDL-C levels can be linked to the hepatic lipase or apo-AI/apo. CIII/apo-AIV/apo-AV gene locus
Con, t • The lack of HDL in the plasma accelerates the development of atherosclerosis, presumably because reverse cholesterol transport or other protective effects of HDL are impaired
Treatments • Drug therapy should be aimed at raising the plasma HDL concentration or lowering the plasma LDL concentration with statins Fibrates (gemfibrozil, fenofibrate) raise • HDL-C levels about 5% to 20%, similar to the effects of statins. Niacin is the most effective drug currently available to raise HDL-C levels