Last lecture summary Flavors of sequence alignment pairwise







 • Match/mismatch score: +1/+0 • Origination/length penalty: – 2/–")









 High gap penalty. Gaps has been inserted only at the beginning and end.")








 Pam 40 Short highly similar alignments")



 • General class of algorithms typically applied to optimization problems. •")







 • Stringency")

 –")





- Slides: 47
Last lecture summary
Flavors of sequence alignment pair-wise alignment × multiple sequence alignment
Flavors of sequence alignment global alignment × local alignment global local align entire sequence stretches of sequence with the highest density of matches are aligned, generating islands of matches or subalignments in the aligned sequences
Evolution of sequences • The sequences are the products of molecular evolution. • When sequences share a common ancestor, they tend to exhibit similarity in their sequences, structures and biological functions. DNA 1 DNA 2 Protein 1 Protein 2 Sequence similarity Similar 3 D structure Similar function Similar sequences produce similar proteins However, this statement is not a rule. See Gerlt JA, Babbitt PC. Can sequence determine function? Genome Biol. 2000; 1(5) PMID: 11178260
Homology • homology, orthology, paralogy • orthologs – from different spcies, posses same function • paralogs – different function in the same organism • How it happens? • orthology – speciation • paralogy – gene duplication • gene duplication – unequal cross-over, chromosome replication, retrotrasposition • The degree of sequence conservation in the alignment reveals evolutionary relatedness of different sequences • The variation between sequences reflects the changes that have occurred during evolution in the form of substitutions and/or indels.
Scoring systems • DNA and protein sequences can be aligned so that the number of identically matching pairs is maximized. A T T G - - - T A – - G A C A T • Counting the number of matches gives us a score (3 in this case). Higher score means better alignment. • This procedure can be formalized using substitution matrix. A Identity matrix T C A 1 T 0 1 C 0 0 1 G 0 0 0 G 1
Scoring DNA sequence alignment • Match score: • Mismatch score: +1 +0 • Gap penalty: – 1 • ACGTCTGATACGCCGTATAGTCTATCT ||||| || |||| ----CTGATTCGC---ATCGTCTATCT • Matches: 18 × (+1) • Mismatches: 2 × 0 • Gaps: 7 × (– 1) Score = +11
Scoring DNA sequence alignment (2) • Match/mismatch score: +1/+0 • Origination/length penalty: – 2/– 1 • ACGTCTGATACGCCGTATAGTCTATCT ||||| || |||| ----CTGATTCGC---ATCGTCTATCT • Matches: 18 × (+1) • Mismatches: 2 × 0 • Origination: 2 × (– 2) • Length: 7 × (– 1) Score = +7
Substitution matrices • Should reflect: • Physicochemical properties of amino acids. • Different frequencies of individual amino acids occuring in proteins. • Interchangeability of the genetic code. • PAM • Manual alignments of 71 groups of very similar (at least 85% identity) protein sequences. 1572 substitutions were found. • These mutations do not significantly alter the protein function. Hence they are called accepted mutations. • Two sequences are 1 PAM apart if they have 99% identical residues. • PAM 1 matrix is the result of computing the probability of one substitution per 100 amino acids. • Higher PAM matrices
Zvelebil, Baum, Understanding bioinformatics. PAM 120 Positive score – frequency of substitutions is greater than would have occurred by random chance. Zero score – frequency is equal to that expected by chance. small, polar Negative score – frequency is less than would have occurred by random chance. small, nonpolar or acidic basic large, hydrophobic aromatic
Selzer, Applied bioinformatics. How to calculate score? 2 substitution matrix
New stuff
Protein vs. DNA sequences • Given the choice of aligning DNA or protein, it is often more informative to compare protein sequences. • There are several reasons for this: • Many changes in DNA do not change the amino acid that is specified. • Many amino acids share related biophysical properties. Though these amino acids are not identical, they can be more easily substituted each with other. These relationships can be accounted for using scoring systems. • When is it appropriate to compare nucleic sequences? • confirming the identity of DNA sequence in database search, searching for polymorphisms, confirming identity of cloned c. DNA
Similarity vs. identity • Similarity refers to the percentage of aligned residues that can be more readily substituted for each other. • have similar physicochemical characteristics and • the selective pressure results in some mutations being accepted and others being eliminated S = [(Ls × 2)/(La + Lb)] × 100 number of aligned residues with similar characteristics total lengths of each sequence
Homology vs. similarity • Two sequences are homologous when they descended from a common ancestor sequence. • Similarity can be quantified: “two sequences share 40% similarity”. • But NOT “two sequences share 40% homology”. Just “two sequences are homologous” • Qualitative statement • And it is a conclusion about a common ancestral relationship drawn from sequence similarity comparison
Gaps • How will I score this alignment? V D S - C Y V E S L C Y • The gaps can’t be inserted freely. • Indels are relatively slow evolutionary processes. • And alignments with large gaps do not make biological sense. • Each gap is penalized – a gap penalty • The gap penalty is an adjustable parameter. • Let’s use the gap penalty equaling to -11. V D S V E S L 4 2 4 -11 C Y 9 7 S = 4 + 2 + 4 – 11 + 9 + 7=15
Gap penalty • Affine gap penalty • different for opening and extending • constant for extending • The gap penalty is high – fewer gaps will be inserted • If you’re searching for sequences that are a strict match for your query sequence, the gap penalty should be set high. • This will retrieve regions with very closely related sequences. • The gap penalty is low – more and larger gaps will be inserted • If you are searching for similarity between distantly related sequences, the gap penalty should be set low.
(A) High gap penalty. Gaps has been inserted only at the beginning and end. Percentage identity = 10% (B) Low gap penalty. More gaps. Percentage identity = 18% Zvelebil, Baum, Understanding bioinformatics.
BLOSUM matrices I • BLOck SUbstitution Matrix by Henikoff and Henikoff, 1992. • They used the BLOCKS database containing multiple alignments of ungapped segments (blocks). • These alignments correspond to the most highly conserved regions of proteins. • Blocks are ungapped sequence motifs. Sequence motif is a conserved stretch of amino acids confering a specific function to a protein. • Any given protein can contain one or more blocks corresponding to its structural/functional motifs.
Blocks . . .
BLOSUM matrices II • Thus the Hanikoffs focused on substitution patterns only in the most conserved regions of a protein. These regions are (presumably) least prone to change. • The substitution patterns of 2000 blocks (block is the whole alignment, not individual sequences within it) representing more than 500 groups were examined, and BLOSUM matrices were generated. • Sequences sharing no more than 62% identity were used to calculate BLOSUM 62 matrix. Short and clear explanation of BLOSUM 62 derivation: Eddy SR. Where did the BLOSUM 62 alignment score matrix come from? Nat Biotechnol. 2004 22(8): 1035 -6. PMID: 15286655.
BLOSUM matrices III • BLOSUM matrices are based on entirely different type of sequence analysis (local ungapped alignment vs. global gapped alignment in PAM) and on a much larger data set than PAM. • All BLOSUM matrices are based on observed alignments. They are not based on extrapolations like PAM. • BLOSUM numbering system goes in reversing order as the PAM numbering system. • The lower the BLOSUM number, the more divergent sequence they represent.
PAM vs. BLOSUM I • However, you may ask a question which particular matrix should be used? • Dayhoff et al. (1978) defined terms protein families and superfamilies. • A protein family is formed by sequences 85% (or greater) identical to each other. • A protein superfamily is defined as sequences related from 30% or greater. • Superfamily may clearly contain many families. • These terms are widely used in contemporary literature, however with different meanings (we’ll come to that later). Guidance in the choice of scoting matrix: Wheeler D. Selecting the right protein-scoring matrix. Curr Protoc Bioinformatics. 2002; Chapter 3: Unit 3. 5. www. nshtvn. org/ebook/molbio/Current%20 Protocols/CPB/bi 0305. pdf
PAM vs. BLOSUM II – PAM • At the time of deriving PAM matrices, most known proteins were small, globular and hydrophilic. If resercher believes his protein contain substantial hydrophobic regions, PAM matrices are not that useful. • Most widely used is PAM 250. • It is capable of detecting similarities in the 30% range (i. e. superfamilies). • Another point of view – PAM 250 provides the best lookback in evolutionary time. • PAM 250 is most effective if the goal is to know the widest possible range of proteins similar to the given protein.
PAM vs. BLOSUM III – PAM • Assume a protein is a known member of the serine protease family. • Using the protein as a query against protein databases with PAM 250 will detect virtually all serine proteases, but also considerable amount of irrelevant hits. • In this case, the PAM 160 matrix should be used. It detects similarities in the 50% to 60% range (Altschul, 1991). • And to find only those proteins most similar (70% - 90%) to the query protein, use PAM 40. • Let’s summarize: • Locate all potential similarities – PAM 250 • Determine if the protein belongs to the protein family – PAM 160 • Determine the most similar proteins – PAM 40
PAM vs. BLOSUM IV – BLOSUM • Most widely used is BLOSUM 62. • BLOSUM 62 appears to be superior to PAM 250 in detecting distant relationships even if the PAM method is updated with current data sets. • BLOSUM 62 is capable of accurately detecting similarities down to the 30% range (superfamilies). • Determine if the protein belongs to protein family – BLOSUM 80 (detects identities at the 50% level) • Determine the most similar proteins – BLOSUM 90
Selecting an Appropriate Matrix Best use Similarity (%) Pam 40 Short highly similar alignments 70 -90 PAM 160 Detecting members of a protein family 50 -60 PAM 250 Longer alingments of more divergent sequences ~30 BLOSUM 90 Short highly similar alignments 70 -90 BLOSUM 80 Detecting members of a protein family 50 -60 BLOSUM 62 Most effective in finding all potential similarities 30 -40 BLOSUM 30 Longer alingments of more divergent sequences <30 Similarity column gives range of similarities that the matrix is able to best detect.
PAM vs. BLOSUM V – battle • Careful information theory analysis showed that the following matrices are equivalent: • PAM 250 is equivalent to BLOSUM 45 • PAM 160 is equivalent to BLOSUM 62 • PAM 120 is equivalent to BLOSUM 80 • Compared to the PAM 160 matrix, BLOSUM 62 is less tolerant to substitutions involving hydrophilic amino acids, and more tolerant to substitutions involving hydrophobic amino acids. • Although both PAM 250 and BLOSUM 62 detect similarities at the 30% level, since BLOSUM uses much wider range of proteins, PAM 250 is actually equivalent to BLOSUM 45 when considering all proteins, not just those that are hydrophilic.
Scoring DNA Alignment • The concept of similarity has little relevance here. • Though transitions (R → R or Y → Y) occur more often than transversions (R → Y or Y → R), this is usually not helpful for sequence alignment. • Instead, concept of identity is used. • Frequencies of mutations are equal for all bases: • match score +5 • mismatch score -4 • gap penalty (usually a parameter) • opening -10 • extending -2
Pairwise alignment algorithms • Dynamic programming • Slow, but formally optimizing • Heuristic methods • Efficient, but not as thorough • Word (also k-tuples) methods • Used in database searches • Dot plot (dot matrix) • Graphical way of comparing two sequences
Dynamic programming (DP) • General class of algorithms typically applied to optimization problems. • Recursive approach. • Original problem is broken into smaller subproblems and then solved. • Pieces of larger problem have a sequential dependency. • 4 th piece can be solved using solution of the 3 rd piece, the 3 rd piece can be solved by using solution of the 2 nd piece and so on…
We want to align two following sequences: ABCDE PQRST If you already have the optimal solution to: A…D P…R then you know the next pair of characters will be either: A…DE P…RS or A…DE P…R- You can extend the match by determining which of these has the highest score.
New best alignment = previous best + local best Best previous alignment Sequence A. . . Sequence B
DP algorithms • Global alignment - Needlman-Wunsch • Local alignment - Smith-Waterman • Guaranteed to provide the optimal alignment. • Disadvantages: • Slow due to the very large number of computational steps: O(n 2). • Computer memory requirement also increases with the square of the sequence lengths. • Therefore, it is difficult to use the method for very long sequences. • Many alignments may give the same optimal score. And none of these correspond to the biologically correct alignment.
Dot plot • Graphical method that allows the comparison of two biological sequences and identify regions of close similarity between them. • Also used for finding direct or inverted repeats in sequences. • Or for prediction regions in RNA that are selfcomplementary and therefore have potential to form secondary structures.
Self-similarity dot plot I The DNA sequence EU 127468. 1 compared against itself. Introduction to dot-plots, Jan Schulz http: //www. code 10. info/index. php? option=com_content&view=article&id=64: inroduction-to-dot-plots&catid=52: cat_coding_algorithms_dot-plots&Itemid=76
background noise gap runs of matched residues
Self-similarity dot plot II The DNA sequence EU 127468. 1 compared against itself. Window size = 16. Linear color mapping Introduction to dot-plots, Jan Schulz http: //www. code 10. info/index. php? option=com_content&view=article&id=64: inroduction-to-dot-plots&catid=52: cat_coding_algorithms_dot-plots&Itemid=76
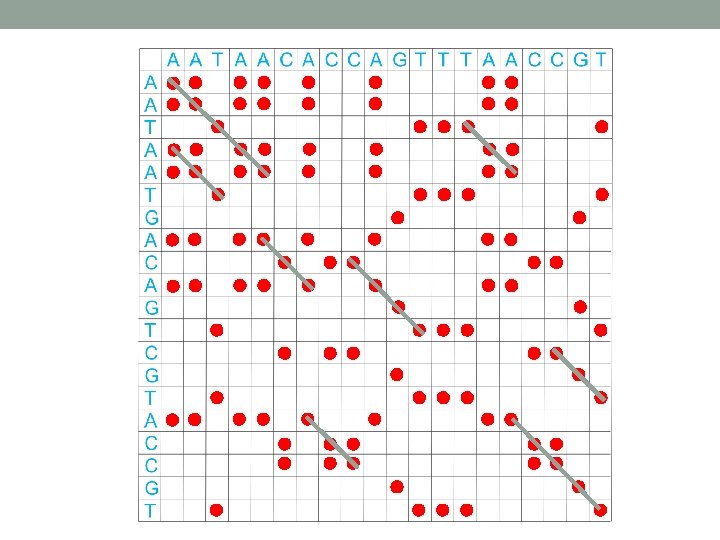
Improving dot plot • Sliding window – window size (lets say 11) • Stringency (lets say 7) – a dot is printed only if 7 out of the next 11 positions in the sequence are identical • Color mapping • Scoring matrices can be used to assign a score to each substitution. These numbers then can be converted to gray/color.
Interpretation of dot plot I 1. Plot two homologous sequences of interest. If they are similar – diagonal line will occur (matches). 2. frame shifts a) mutations gaps in diagonal b) insertions shift of main diagonal c) deletions shift of main diagonal http: //ugene. unipro. ru/documentation/manual/plugins/dotplot/interpret_a_dotplot. html
Interpretation of dot plot II • Identify repeat regions (direct repeats, inverted repeats) – lines parallel to the diagonal line in self-similarity plot • Microsattelites and minisattelites (these are also called low-complexity regions) can be identified as “squares”. • Palindromatic sequences are shown as lines perpendicular to the main diagonal. • Plaindromatic sequence: V ELIPSE SPI LEV Bioinformatics explained: Dot plots, http: //www. clcbio. com/index. php? id=1330&manual=BE_Dot_plots. html
Repeats in dot plot minisattelites self-similarity dot plot of NA sequence ofhuman LDL receptor window 23, stringency 7 direct repeats inverted repeats from the book Bioinformatics, David. M. Mount,
Interpretation of dot plot – summary http: //www. code 10. info/index. php? option=com_content&view=article&id=64: inroduction-to-dot-plots&catid=52: cat_coding_algorithms_dot-plots&Itemid=76
Dot plot of the human genome A. M. Campbell, L. J. Heyer, Discovering genomics, proteomics and bioinformatics
Dot plot rules • Larger windows size is used for DNA sequences because the number of random matches is much greater due to the presence of only four characters in the alphabet. • A typical window size for DNA is 15, with stringency 10. For proteins the matrix has not to be filtered at all, or windows 2 or 3 with stringency 2 can be used. • If two proteins are expected to be related but to have long regions of dissimilar sequence with only a small proportion of identities, such as similar active sites, a large window, e. g. , 20, and small stringency, e. g. , 5, should be useful for seeing any similarity.
Dot plot advantages/disadvantages • Advantages: • All possible matches of residues between two sequences are found. It’s just up to you to choose the most significant ones. • Readily reveals the presence of insertions/deletions and direct and inverted repeats that are more difficult to find by the other, more automated methods. • Disadvantages: Most dot matrix computer programs do not show an actual alignment. Does not return a score to indicate how ‘optimal’ a given alignment is (no statistical significance that could be tested).