DNA Sequences Analysis Hasan Alshahrani CS 6800 Statistical

 is very useful statistical model for molecular biology although")








")










")



- Slides: 26
DNA Sequences Analysis Hasan Alshahrani CS 6800 • Statistical Background : HMMs. • What is DNA Sequence. • How to get DNA Sequence. • DNA Sequence formats. • Analysis methods and tools. • What is next ?
HMMs Hidden Markov Model (HMM) is very useful statistical model for molecular biology although it was aimed to be used for speech recognition purposes. HMM can be used as a statistical profile for a protein family (DNAs) and hence used to search a database for other similarities or family members. Q 1 : How can HMMs be used in DNA analysis?
To calculate the probability of the sequence ACTTCG, we multiply the probabilities; where the probability is the conditional probability that a certain nucleotide appears in a position, given that a specific nucleotide was in the previous position: P (ACTTCG…. ) = P 1(A) * P 2(C|A) * P 3(T|C) * P 4(T|T) * P 5(C|T) * P 6(G|C)…………
In more formal way , HMM cannot be observed directly but we can infer the hidden state qt from a random observation Yt
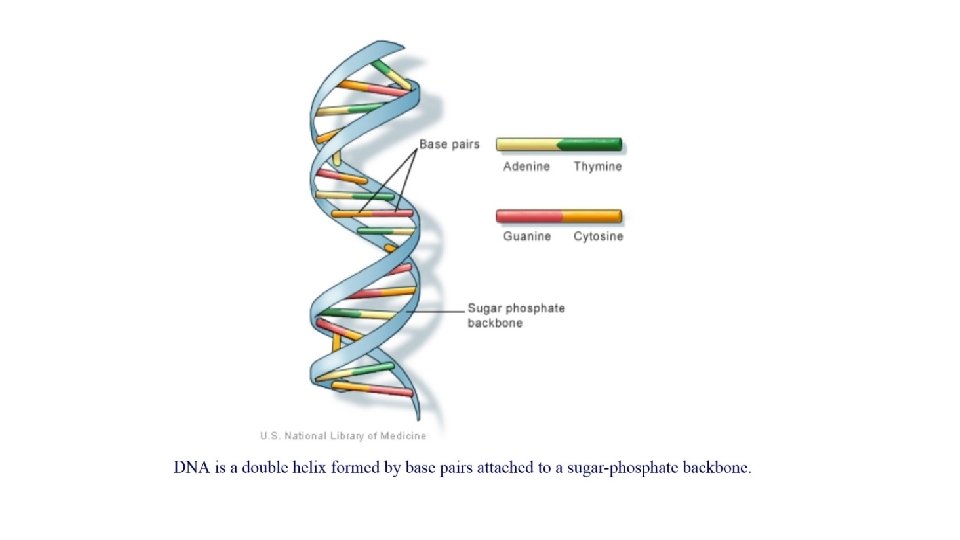
What is DNA sequence ? • DNA consists of two long interwoven strands that form the famous “double helix”. Each Strand is built from a small set of molecules called nucleotides. • Often the length of double-stranded DNA is expressed in the units of basepairs (bp), kilobasepairs (kb), or megabasepairs (Mb), so that this size could be expressed equivalently as 5 X 10 ^6 bp, 5000 kb, or 5 Mb • Collectively, the 46 chromosomes in one human cell consist of approximately 3 X 10^9 bp of DNA
How to get DNA sequence • By using chemical methods for determining the order of the nucleotide bases: Adenine, Guanine, Cytosine, and Thymine - in a molecule of DNA • Used in many fields and applications such as Forensics and biological systems • why don’t we use the powerful text searching algorithms and tools to search DNA databases?
DNA can be sequenced by a chemical procedure that breaks a terminally labelled DNA molecule partially at each repetition of a base. DNA Sequencing can be done by different methods : 1. Maxam-Gilbert sequencing 2. Chain-termination methods 3. Dye-terminator sequencing 4. Automation and sample preparation 5. Large scale sequencing strategies Q 2: Name four of DNA Sequencing methods
Example : a chain termination method A DNA sequencing printout. The sequence is represented by a series of peaks, one for each nucleotide position. In this example, a red peak is an A, blue is a C, orange is a G, and green is a T.
DNA Sequence formats: • Plain sequence format • EMBL format • FASTA format • GCG-RSF (rich sequence format) • Gen. Bank format • IG format
FASAT Format : • FASTA format is the standard format in the field of bioinformatics to represent either nucleotide sequences or peptide sequences. • This format is single-letter code and it allows sequence names and comments • FASAT consists of a single-line description at the beginning followed by sequence data in multiple lines. • The length of the each chunk (line) of the sequence must not exceeds 80 characters. • Sequence identifiers are defined by a standard called NCBI Q 3: what is FASAT format?
NCBI Data Base: • National Centre for Biotechnology Information (www. ncbi. nlm. nih. gov) is sequence database in US maintain a huge collection of DNA and protein sequences. • Each sequence in NCBI is stored in a separate record with a unique identifier called accession. • Example : By accessing the NCBI website and using this accessing NC_001477, we can retrieve the DNA sequence for Dengue virus that causes Dengue fever
NCBI cont…. . The database query can be done either directly from the website or by using the R functions choosebank() and query()
Analysis methods The analysis fall into 5 main methods : • Knowledge-based single sequence analysis. • Pairwise sequence comparison. • Multiple sequence alignment. • Sequence motif discovery in multiple alignments. • Phylogenetic inference. Q 4: What are the main methods of DNA sequence analysis ?
Analysis methods: alignment • Alignment: to compare a sequence with sequences that have already been reported and stored in a database. • Alignment can be global and local • Local alignments: reveal regions that are highly similar, but do not necessarily provide a comparison across the entire two sequences. • The global approach compares one whole sequence with other entire sequences.
Alignment Examples:
Alignment Tools : BLAST • The most common local alignment tool is BLAST (Basic Local Alignment Search Tool) developed by Altschul et al. (1990. J Mol Biol 215: 403) “BLAST is a set of algorithms that attempt to find a short fragment of a query sequence that aligns perfectly with a fragment of a subject sequence found in a database. ” • That initial alignment must be greater than a neighborhood score threshold (T) , the fragment is then used as a seed to extend the alignment in both directions… Which means BLAST algorithm breaks the query into short words of a specific length
Joshua Naranjo Q 5: what is BLAST algorithm ? State its steps.
Can R Help ? • Yes. • It has so many useful packages to process DNA Sequences. • It can be used to access BLAST as well.
Examples : DNA sequence Composition 1. GC fraction: GC content is one of the fundamentals properties of a genome sequence, which is the percentage of Gs and Cs ((GC)s). We can do that by two ways: • lengthy one is to use the statistics to calculate the percentage of GC with respect to the whole string. • The other way is to use function GC () from the R package Seqin. R, and we will go with this option as shown below
2. DNA words: It the same idea of knowing the frequency of some nucleotides such as A or G but with longer words like “AA” or “CA”. Those can be 2 nucleotides such as “GC”, 3 nucleotides like “AAA” or 4 nucleotides long and so on. An example of 3 nucleotides words is shown below:
3. To find the score for the optimal global alignment between the sequences ‘GAATTC’ and ‘GATTA’, we type:
4. Comparing two sequences using a dotplot()
Is it that easy ? No • It is not simply give the sequences to R and get the results. • It is an art which need a degree of skills. • Fitting the sequences to be compared to a form that reflects some shared quality. For example: -How they look structurally, -How they evolved from a common ancestor, or -Optimization of a mathematical construct
What is next ? Are we monkeys ?
References: 1. 2. 3. 4. 5. 6. 7. 8. http: //www. garlandscience. com/res/pdf/9780815365099_ch 02. pdf http: //library. umac. mo/ebooks/b 28050393. pdf https: //courses. cs. washington. edu/courses/cse 527/00 wi/lectures/roottr. pdf http: //www. lancaster. ac. uk/pg/nemeth/Hidden%20 Markov%20 Models%20 with%20 Applications%20 to%20 D NA%20 Sequence%20 Analysis. pdf https: //www. ndsu. edu/pubweb/~mcclean/plsc 411/Blast-explanation-lecture-and-overhead. pdf http: //www. cs. ru. ac. za/research/g 07 V 3343/deliverables%5 CShort%20 Paper%5 CSpecies%20 Identification%2 0 through%20 DNA%20 String%20 Analysis%20 -%20 Summary. pdf http: //a-little-book-of-r-for-bioinformatics. readthedocs. org/en/latest/src/chapter 4. html https: //www. bioconductor. org/packages/3. 3/bioc/vignettes/DECIPHER/inst/doc/Art. Of. Alignment. In. R. pdf