Srovnn sekvenc zkladn vzorce a a 1 a



 Delece a inzerce Komprese a expanze Transpozice I")



 Inicializace skórovací matice (0 nebo 1) 2) Sumace -nalézt")
















 Je stvořena populace individualit - individua jsou charakterizována a")

 Výběr individuí pro novou generaci rodičů - v originálním genet. algoritmu se zavrhnou")










- Slides: 38
Srovnání sekvencí - základní vzorce a= a 1 a 2 a 3………. . a 100 b= b 1 b 2 b 3………. . b 100 Euklidovská vzdálenost City Block vzdálenost Hammingova vzdálenost # počet odlišných pozic
a= 10101 b= 001110100 Euklidovská vzdálenost = City Block vzdálenost 1+0+0+0+0+1=3 Hammingova vzdálenost = 3
Dvě struktury jsou homologické tehdy, mají li společného evolučního předka, nebo mají li podobnou strukturu + funkci. Struktury mají vysoký stupeň homologie jsou li mezi nimi relativně malé rozdíly. Jsou určité makromolekuly homologické? Jaká část jedné molekuly je homologická k jaké části druhé molekuly? Jaké dvě makromolekuly mají typicky vysoká stupeň homologie?
Jak se sekvence liší? Substituce (výměna) Delece a inzerce Komprese a expanze Transpozice I N D U S T R Y I N T E R E S T
Alignment nebo shoda INDUST R Y IN TEREST INDUSTRY INSTRY INSTRS INSTERES INTEREST Delete D Delete U Subst Y by S Insert E Delete S Insert T
Různé analýzy stejného páru W | W A T R N E | E I W | W A | I T | N E | E R W A T E R W I N E W A T E W I N E R R
Algoritmus - Dynamické programování - podobný koncept jako „ dot matrix“ Aplikována na biologické sekvence S. B. Needleman & C. D. Wunsch. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48: 443 -453 (1970)
Základní kroky dynamického programování 1) Inicializace skórovací matice (0 nebo 1) 2) Sumace -nalézt maximální počet shod který může být získán počínaje libovolnou pozicí a pokračováním „vpřed“ 3) Traceback k nalezení maximálního alignmentu
Sumace: 1. Start v pravém dolním rohu 2. Pohyb nahoru a vlevo o jednu pozici 3. Nalezení největší hodnoty buď, v a) v segmentu řádku počínajícím jeden pod aktuální pozicí a pokračováním vpravo, nebo b) v segmentu sloupce počínajícím jeden vpravo od aktuální pozice a pokračováním dolů 4. Připočtení této hodnoty k hodnotě aktuálního políčka 5. Zopakování kroku 3 a 4 pro všechna políčka vlevo od aktuálního řádku a nahoru od aktuálního sloupce dokud se nedospěje k levému okraji matice. 6. Pokud nejsme v levém horním rohu, pokračovat 2
Aplikace Hidden Markova Modelu na proteiny stejné core všech 20 aminokyselin - karboxylová kyselina - aminoskupina sekvence – primární struktura
CGGSLLNAN--TVLTAAHC CGGSLIDNK-GWILTAAHC CGGSLIRQG--WVMTAAHC CGGSLIREDSSFVLTAAHC Primární struktura 4 příbuzných proteinů CGSLIREDWVLTAAHC Možný společný předek
Jednoduchý statistický profile
Pravděpodobnost výskytu CGGSV 0. 8 * 0. 4 * 0. 8 * 0. 6 * 0. 2 =. 031 Tímto výpočtem získáváme score pro určitou sekvenci. (Transformace do logaritmické funkce) loge(0. 8)+loge(0. 4)+loge(0. 8)+loge(0. 6)+loge(0. 2) = -3. 48
Hidden Markov Model je druh dynamického statistického profilu Má komplexnější topologiii HMM lze vizualizovat jako stroj finitních stavů Stroj finitních stavů – pohybuje se skrze série stavů a produkuje výstupní stav ať už se stroj nachází v určitém stavu, nebo se pohybuje mezi nimi. HMM generuje sekvenci proteinu emisí AA při průchodu sériemi stavů. Každý stav je charakterizován tabulkou emisních pravděpodobností podobnýcj jako v profilu. Existují i tranzitní pravděpodobnosti.
HMM – základní schéma
delete insert match Možný HMM pro sekvenci ACCY. Protein je representován jako sekvence pravděpodobností. Čísla ukazují pravděpodobnosti, že se která aminokyselina nachází v danném stavu. Čísla u šipek ukazují pravděpodobnosti přechodu mezi stavy.
Libovolná sekvence může být representována jedinečnou cestou v HMM. Pravděpodobnost určité sekvence je určena jako součin emisních a transitních pravděpodobností podél určité trajektorie (cesty) ACCY 4 *. 3 *. 46 *. 97 *. 5 *. 015 *. 73 *. 01 * 1 = 1. 76 x 10 -6. loge(. 4) + loge(. 3) + loge(. 46) + loge(. 97) + loge(. 5) + loge(. 015) + loge(. 73) +loge(. 01) + loge(1) = -13. 25 Výpočet je jednoduchý je li známa cesta. Ve skutečném modelu existuje mnoho různých cest generující téže sekvenci. Proto přesná pravděpodobnost sekvence je suma pravděpodobností přes všechny možné stavové trajektorie.
Výpočet nejlepší cesty: - Viterbův algoritmus - forwarding algoritmus Problém ACCY: stavy: M – match, I – insertion, D – deletion 1)Pravd. že A je generováno jao stav I 0 je vypočteno a vneseno do matice 2)Pravd. že C je emitováno do stavu M 1 a do stavu I 1 je vneseno do matice jako C a I 1/M 1 3)vypočte se max (I 1/M 1) 4)pointer je posunut od vítěze do stavu I 0 5)opakuje se 2 -5 dokud se matice nenaplní
Matice Viterbiho algoritmu
Význam score: Model je generalizací jak jsou AA distribuovány v určité grupě příbuzných sekvencí. Score tedy znamená příslušnost k danné třídě. Lokální versus globální scoring.
Problémy: Vybudování setu pro HMM, je třeba odhadnout emisní koeficienty. K tomu je třeba série příbuzných testovacích sekvencí. Pokud je stavová trajektorie známa, je možné vypočítat jednotlivé pravděpodobnosti. V opačném případě je nalezení nejlepšího modelu pro danou testovací sadu problémem který nemá řešení v uzavřené formě.
Vážení sekvencí: malá skupina vysoce podobných sekvencí může vnést do modelu nechtěnou závislost. řešení: - vážení sekvencí
I 0=I 1+I 2 I 1=I 2 I 2=I 3+I 4 I 3=I 5+I 6 I 5=I 6 I 4=I 7+I 8 I 7=I 8 I 1=I 2=. 5 * I 0 I 3=I 4=. 25*I 1 I 5=I 6=I 7=I 8=. 125 * I 1
Genetický algoritmus The so-called genetic algorithm is a heuristic method that operates on pieces of information like nature does on genes in the course of evolution. Individuals are represented by a linear string of letters of an alphabet (in nature nucleotides, in genetic algorithms bits, characters, strings, numbers or other data structures) and they are allowed to mutate, crossover and reproduce. All individuals of one generation are evaluated by a fitness function. Depending on the generation replacement mode a subset of parents and offspring enters the next reproduction cycle. After a number of iterations the population consists of individuals that are well adapted in terms of the fitness function.
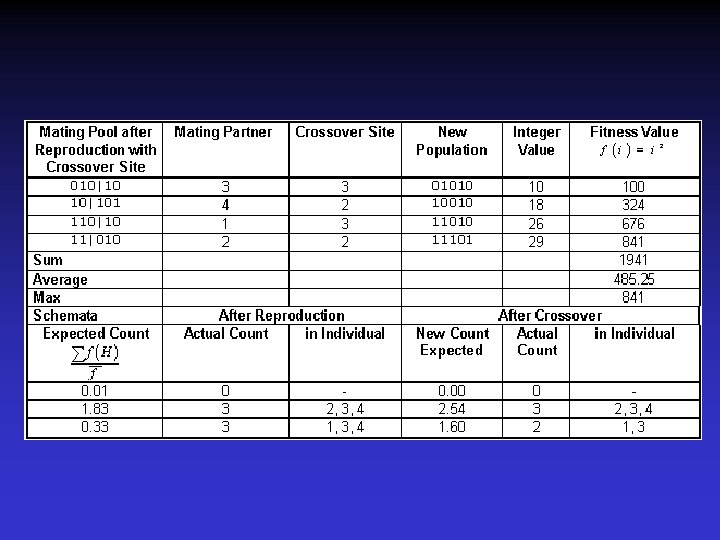
Základní popis genetického algoritmu 1) Je stvořena populace individualit - individua jsou charakterizována a vyjádřena jako sekvence bitů. (obecně – řada) - je definována tzv. fittness funkce. je definována tak, že vezme jako vstup individuum a poskytne jako výstup číslo nebo vektor který udává kvalitu individua - určí se hierarchie individuí podle fittness funkce 2) Provede se ohodnocení všech individuí v první populaci 3) Vytvoří se nová individua. Reprodukční schopnost individuí je proporcionální jejich hierarchii v danné populaci. Zahrnuje následující operace
Mutace Variace Křížení
4) Výběr individuí pro novou generaci rodičů - v originálním genet. algoritmu se zavrhnou rodiče a pouze individua z nové generace mohou tvořit příští rodiče - upravovaný GA uvažuje pro zhodnocení celou populaci včetně rodičů. Do další generace jsou selektováni fittness funkcí. (tzv. elitářská výměna) 5) opakuj kroky 2 až 4 dokud není dosaženo požadované vlastnosti, nebo dokud neproběhne předepsaný počet iterací Matematické základy GA položil J. H. Holland v tzv. „schemata theorem“ -schema je generalizací nebo částí individua
0101001010111010101 a 0101101010010111000111010111 může být sumarizováno schematem: 0101#010100101#10#011101#10101#1 Očekávané množství výskytu určitého schematu v čase t+1
Úloha: největší druhá mocnina integer < 32
Evoluční strategie: jde o optimalizační problém stejně jako u GA Rozdíly: -ES byla vytvořena jako optimalizační funkce -reprodukce v GA je proporcionální fittness funkci, nikoli v ES -GA činí rozdíly mezi genotypem a fenotypem, ES nikoli -v ES rodiče i potomci kompetují o přežití, nikoli v orig. GA -mutace je řídící silou u ES zatímco pro GA je to křížení
Hydropathy/Hydrophilicity/ Hydrophobicity • Hydropathy & Hydrophobicity – stupeň ukazující “water hating” či “water fearing” • Hydrophilicity – stupeň ukazující “water loving”
Hydropathy/Hydrophilicity/ Hydrophobicity Analýza Cíl: Nalézt kvantitativní popis stupně expozice proteinu do vodného prostředí Východisko: Tabulka expozic jednotilých aminokyselin
Hydrophobicity/Hydrophilicity Tables • Popisuje pravděpodobnost pro každou aminokyselinu, že bude nalezena ve vodném prostředí • Používaná kriteria – Kyte-Doolittle hydropathy – Hopp-Woods hydrophilicity – Eisenberg et al. normalizovaná consensuální hydrophobicita
Kyte-Doolittle hydropathy
Hydrophilicity Plot - Příklad okno 7 AA Tento plot je pro tubulin, rozpustný cytoplasmatický protein. Regiony s vysokou hydrophilicitou jsou pravděpodobně exponovány do solventu (cytoplasmy), zatímco hydrophilní jsou pravděpodobně uvnitř nebointeragují s jinou částí proteinu
Amphiphilicity/Amphipathicity K nalezení takové sekvence hledáme oblasti kde se střídají krátké úseky nabitých aminokyselin s kratkými úseky hydrofobních v opakované délce která koresponduje s periodou ve struktuře