A haemostasis s betegsgeinek genetikai vonatkozsai RKLETES RENDELLENESSGEK

A haemostasis és betegségeinek genetikai vonatkozásai

ÖRÖKLETES RENDELLENESSÉGEK • Thrombofíliák • Koagulopátiák • A thrombocyták veleszületett rendellenességei

THROMBOPHILIA: FOKOZOTT THROMBOSIS HAJLAMMAL JÁRÓ ÁLLAPOTOK • Veleszületett • Szerzett
 kialakuló trombózis Szokatlan helyen")
TROMBOFÍLIÁRA HÍVJA FEL A FIGYELMET • • Fiatal korban (50) kialakuló trombózis Szokatlan helyen kialakuló trombózis Recidíváló trombózis Enyhe provokáló tényező mellett kialakuló trombózis • Habituális vetélés, koraszülés, halvaszülés

ÖRÖKLETES TROMBOFÍLIÁK • genetikai hiba • Családi halmozódás • Általában vénás trombózisok gyakorisága nő meg • Gyakori eltérések enyhe trombózis hajlammal járnak • Ritkábbak súlyosabbal

GYAKORIBB ÖRÖKLETES TROMBOFÍLIÁK • • • VF Leiden mutáció Aktivált protein C rezisztencia Prothrombin gén mutáció Hyperhomocysteinaemia Emelkedett FVIII szint

RITKÁBB ÖRÖKLETES TROMBOFÍLIÁK • • • Antithrombin III defektus Protein C rendszer rendellenessége Protein S defektus VF Leiden mutáció - homozygota Prothrombin gén mutáció - homozygota

TÖRTÉNETI ÁTTEKINTÉS ● ● ● 1965. Egeberg, AT defektus első familiáris trombofília leírása 1981. protein C defektus 1986. Magyarországon az első protein C hiányos család, Domján 1984. protein S hiányos család 1993. APC rezisztencia, Dahlback 1994 FV Leiden mutáció, Bertina

ÖRÖKLETES THROMBOPHILIÁK • • Antithrombin III defektus Protein C rendszer rendellenessége Protein S defektus Aktivált protein C rezisztencia VF Leiden mutáció Prothrombin gén mutáció Hyperhomocysteinaemia „Sticky platelet syndrome” • • Dysfibrinogenaemia Thrombomodulin mutációk Plasminogén defektus Heparin kofaktor II hiány Szöveti faktor út gátló (TFPI) hiány XII F részleges hiánya Genetikusan determinált emelkedett VIII F szinttel járó állapot Az ok általában 80 -90 %-ban tisztázható

TERMÉSZETES ANTIKOAGULÁNSOK • Az alvadási folyamat inhibitorai • Szerep: a trombus képződés az érsérülés helyén lokalizált maradjon

Antithrombin III – heparin rendszer • AT III szerepe: serin proteáz inhibitor • Aktivált alvadási faktorok irreverzibilis inaktiválása • Xa / IIa / Ixa / XIa inaktivációjában vesz részt • Hatását a heparin, és az EC felszínén lévő heparán szulfát fokozza

Antithrombin III – heparin rendszer • Fontos: AT hiányában a heparin kevésbé hat!

AT – heparin – trombin komplex

Antithrombin III defektus • Az első felismert örökletes thrombophilia • Egeberg 1965, első ATIII hiányos család • Általában nem teljes hiány, csak kb 50 %-os csökkenés • Hiány, csökkenés, funkcionális eltérések • Mutációk heterogén csoportja, okozhat funkció csökkenést is • Lehet szerzett is (pl. májbetegségekben) • Autoszomális domináns öröklődés • kódoló gén az 1. kromoszómán van

Antithrombin III defektus § Felosztás: § I. mennyiségi csökkenés § § § homozygota nem életképes, iu. meghal heterozygota: 50 %-os ag csökkenés 20 %-os trombózis rizikó Már fiatal korban MVT, OAK szedés mellett már az első 1 -2 hónap során MVT

Antithrombin III defektus § II. Funkcionális eltérés, normális ag szint § antigén szint normális, vagy enyhén csökkent, § kóros működés, heterogén csoport: attól függően, hogy hol van a mutáció reaktív, vagy heparin-kötő receptort érinti § Reaktív kötőhely mutációi: trombint nem képest közömbösíteni ritka, 20 x rizikó § Heparin kötőhely mutációi: ritka homozygota mutáció: már gyermekkorban súlyos MVT heterozygota: gyakran tünetmentes

Antithrombin III defektus • 20 x thrombosis rizikó • Más genetikai zavarral társulva nagyobb rizikó • Ritka, MVT 1%-a, I. típus: egészséges populáció 0, 02% I-II. típus: 0, 16 %
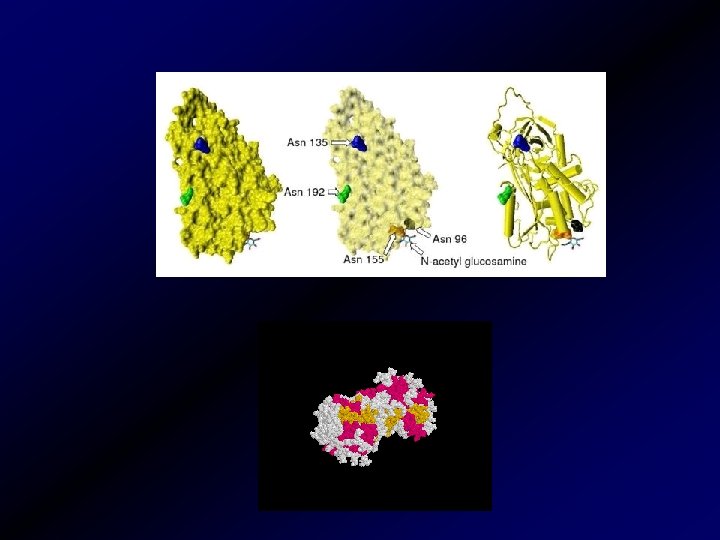

A PROTEIN C RENDSZER • EC-trombomodulin-trombin kapcsolódás • trombin prokoaguláns hatása elvész • PC aktíválása • APC keletkezése (szerin proteáz) • PS, Ca, PL jelenlétében Va, VIIIa inaktiváció • Kevesebb trombin keletkezik • alvadásgátlás

APC REZISZTENCIA • • Dahlback, 1993 A leggyakoribb kongenitális trombofília Szerzett APC: a. PL jelenléte esetén Kimutatás: egészséges: + APC a. PTI 2 x megnyúlása APC rezisztens beteg: a. PTI nem nyúlik meg • Háttérben legtöbbször FV mutációja FV kóros, rezisztens az APC-vel szemben

FV - LEIDEN • • • • 1994 -ben Bertina írta le; University of Leiden FV pontmutációja Kódoló gén az 1. kromoszómán Missence mutáció: 1961 -es pozícióban G-A báziscsere G 1691 A 506 arg glu aminosav cserét eredményez A mutáció az APC kapcsolódási pontjánál van Mutáns FV lassabb inaktiválódás fokozott trombózis készség FVIII hasítása zavartalan FV-t is hasítja a másik 2 helyen, csak lassabb az inaktíválás enyhe trombózishajlam fokozódás Főleg más tényezők együttes jelenléte esetén Jelölés: FVR 506 Q, FVLeiden Autoszóm domináns öröklődés menet Ez felel az APC rezisztencia 95 %-áért

FV – LEIDEN GYAKORISÁG – TROMBÓZIS RIZIKÓ • Homozygota: 80 x rizikó • Heterozygota: 7 x rizikó, általában egyéb tényező jelenléte esetén • Gyakoriság: kaukázusi populációban: 6 -10 % MVT-ben: 20 % fiatal, recidív trombózis: 40 % • Habituális vetélés hátterében • Artériás trombózis ritkább, de: fiatal, dohányos, AMI-n átesett nők: 32 x rizikó, kombinált rizikó: terhesség, szülés, műtét • Hemofília A: kedvező hatás, • részben kompenzálja a VIII f hiányt, • trombin képződés

Protein C deficiencia • Griffin, 1981 • Első magyar PC hiányos család: Domján, és mtsai: 1986 • 2 fenotípus • 1. típus: ag és aktivitás is gyakoribb • 2. típus: ag norm. , aktivitás ritkább • Kódoló gén a 2. kromoszómán • Autoszómális domináns öröklődés • Homozygota. PC < 1 % heterozygota: PC 50 % • Gyakoriság: normál populáció: 0, 3 %, MVT: 3, 2 %

Klinikum • Homozygota: purpura fulminans neonatorum heterozygota: 8 -10 x trombózis rizikó terhesség, OAK, egyéb defektus jelentős trombózis hajlam fokozódás • Kumarin okozta bőrnekrózis heparinnal átfedés kis kumarin kezdő dózis PC koncentrátum, FFP • Protein C és gyulladás sepsis – purpura fulminans

Protein S deficiencia • • • Comp és Esmon, 1984 PS szerepe: PC aktiválás PS szabad formája aktív (40%) C 4 b-hez kötve (60%) (szerep: apoptosisban) PS defektus formái: 1. ag és funkció 2. ag norm. , csak a funkció (ritkább) 3. csak a szabad frakció csökkkent Gyakoriság: PC deficienciához hasonló súlyos: ag < 1 %, purpura fulminans • Mutációk többsége pontmutáció • Kumarin nekrózis hátterében

Prothrombin gén mutáció • Poort, 1996 • 2. leggyakoribb ismert trombofíliát okozó tényező • Missence mutáció, G A csere FII G 20210 A • Plazma FII szintje nő (> 115%) mechanizmus ismeretlen heterozygota • Homozygota: normál homozygota • Heterozygota: 3 x rizikó • Prevalencia: 2% MVT: 6 -10 %

Hyperhomocysteinemia • Leggyakoribb ok: cisztation- -szintetáz hiány ritka, homozygóta súlyos trombózis hajlam • Metilén-tetrahidrofolát reduktáz hiány (MTHFR) • homozygóta súlyos • Artériás és vénás trombózis is gyakoribb • Fiatalokon atherosclerosis • Lehet szerzett is (B 12, B 6, folsav hiány)

Emelkedett VIII faktor szint • Egészséges populáció 10 %-ában • > 150% • 5 x trombózis rizikó

A TROMBÓZIS RIZIKÓ MÉRTÉKE • • Heterozygota FVLeiden: Homozygota FVLeiden: Heterozygota protein C deficiencia: FII G 20210 A homozygota FII G 20210 A heterozygota AT hiány AT homozygota FVL (7 x) + OAK (4 x) 7 x 80 x 10 -15 x 3 -6 x 10 x 28 x

ÖRÖKLETES KOAGULOPÁTIÁK VÉRZÉS TROMBÓZIS

Von Willebrand betegség • A leggyakoribb örökletes vérzékenység • A v. Wf óriás glikoprotein • Szerep: • FVIII stabilizálás • Thrombocyta adhézió • Hiányában: • Primer és secunder hemosztázis is zavart szenved

Von Willebrand betegség • Autoszómális domináns öröklődés menet • v. Wf-t kódoló gén a 12. kromoszómán • MK, EC, placenta sejtjeiben expresszálódik • Mutáció következtében nem termelődik fehérje, vagy kóros fukció • A 22. kromoszómán kisebb méretű pseudogén

Gyakoriság • Kezelést igénylő betegség: 1/10. 000 • A hordozók többsége tünetmentes • 1. típus (70%): mennyiségi csökkenés általában heterozygota • 2. típus: funkció zavar, heterogén csoport, heterozygoták • 3. típus: v. Wf hiány, súlyos vérzékenység • 2 -3. típusnak autoszomális recesszív formája is van

HEMOFÍLIÁK § Hemofília A FVIII hiány § hemofília B FIX hiány, diszfukció § FVIII és F IX génje az X kromoszómán § Nők hordozók (heterozygoták) § Férfiak betegednek meg (hemizygoták) § Ritkán a nők is lehetnek betegek (gén inaktiváció, mozaicizmus, hordozó anya-beteg apa gyereke) § Klinikailag nem különíthető el

A BETEGSÉG SÚLYOSSÁGA • • A faktor szinttől függenek Súlyos: < 1% Kp: 1 -5 % Enyhe: >5%

• Szerep: • FX aktiválása

ÖRÖKLŐDÉS
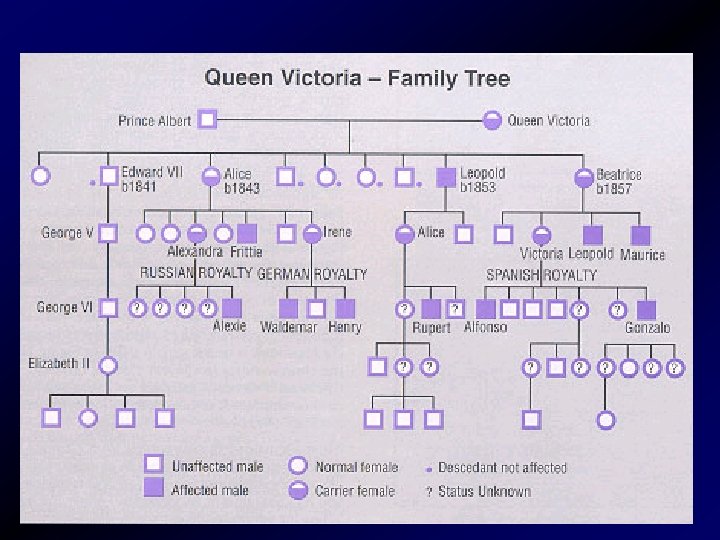
")
„KIRÁLYI BETEGSÉG” Viktória királynő Alekszisz (II. Miklós cár fia)

HEMOFÍLIA A • • • Gyakoriság: 1: 10. 000 5 x gyakoribb, mint „B” 30 %-ban új mutáció Genetikai háttér: Pontmutáció, inverzió, deléció Heterogén eltérések Súlyos esetek 50 %-a: intron 22 inverzió Southern blott-tal kimutatható Többi eltérés indirekt molekuláris genetikai vizsgálatokkal

HEMOFÍLIA B • Kimutatás: indirekt genetikai vizsgálatok FIX génje rövidebb, szekvencia analízis

• Hordózó állapot, prenatális diagnosztika lehetséges

KLINIKUM • Izületi bevérzések

• Nagy, lapszerint vérzések Epiduralis hematoma „re bleeding”
")
Diagnózis • a. PTI megnyúlt • Normál plazmával korrigálható (ha nem: gátlótest)

Differenciál diagnózis • • „A”: v. WD FXI hiány Szerzett gátlótestes hemofília • • • „B” K vitamin hiány Kumarin kezelés Májbetegség APS FIX elleni antitest

A jövő terápiája • gén terápia • Az alvadási faktor génjét májsejtekbe viszik be •

A THROMBOCYTOPATHIÁK VELESZÜLETETT BETEGSÉGEI • • • Normális thrombocytaszám Megnyúlt vérzési idő Klinikailag bőr, nyálkahártya vérzések Ritka betegségek Általában autoszomális recesszív öröklődésű géndefektusok • Dg: thrombocyta funkciós labor vizsgálatok • Vérlemezkék morfológiai eltérései
 Thrombocyta aggregáció Elektromikroszkópos")
VIZSGÁLATOK • • • Vérzési idő Kóros morfológia (óriás, kicsi thrombocyták) Thrombocyta aggregáció Elektromikroszkópos vizsgálat Thrombocyta membrán receptor vizsgálata PFA-100 – platelet function analyzer készülék

AZ EGYES KÓRKÉPEKBEN • • Az Adhézió Aggregáció Szekréció Intracelluláris szignál átvitel Prokoaguláns aktivitás zavara

Az adhézió zavara • • Bernard-Soulier szindróma Óriás thrombocyták GPIb-IX-V komplex képzési zavara A kóros T nem tud kitapadni a subendoteliális kollagénhez • Pseudo-von-Willebrand –betegség • GPIb receptor fokozottan köti a v. Wf-t, a keringésben csökken a v. Wf szint • Kollagén receptor hiány GPVI és GPIa-IIa hiánya

Az aggregáció zavara • Glanzmann thrombasthenia autoszómális recesszív öröklésmenet aggregáció hiánya súlyos vérzések GPIIb-IIIa receptorokat kódoló gének mutációi

A szekréció zavarai • Enyhe, vagy mérsékelt súlyosságú vérzések • Thrombocyta granulumok hiánya, szekréciós mechanizmus rendellenességei • Gray platelet szindróma granulum hiánya – 40 eset • Quebec thrombocyta betegség • Storage-pool disease • Hermansky-Pudlak szindróma • Chediak-Higashi szindróma fvs kemotaktikus és baktericid aktivitás károsodott, infekciók, albinizmus • Wiskott-Aldrich szindróma X-hez kötött, ekzema, immundeficiencia, kis thrombocyták

Wiskott-Aldrich Bernard Soulier Gray platelet

KÖSZÖNÖM MEGTISZTELŐ FIGYELMÜKET
- Slides: 56