MUCOPOLYSACCHARIDOSISMPS Dr NZIOKI CM CONSULTANT ENDOCRINOLOGIST MACHAKOS LEVEL


 are hereditary progressive disorders caused by defective catabolism of sulfated")

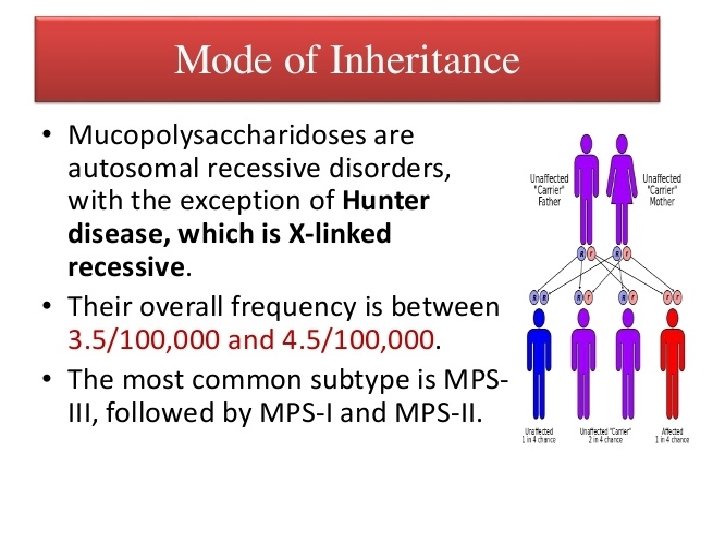




 • • Autosomal recessive mode of inheritance Due")







- Slides: 21
MUCOPOLYSACCHARIDOSIS-MPS Dr. NZIOKI CM CONSULTANT ENDOCRINOLOGIST MACHAKOS LEVEL 5 HOSPITAL
Outline • • • Introduction Mode of inheritance Classification Clinical presentation Diagnosis Management of MPS
INTRODUCTION • Mucopolysaccharidosis (MPS) are hereditary progressive disorders caused by defective catabolism of sulfated components of connective tissue- glycosaminoglycans (GAG’s) • The major GAGs are: v dermatan sulfate (DS) v heparan sulfate(HS) v keratan sulfate(KS) v. Chondroitin -4 - sulfate v. Hyaluronan
Introduction conti’ • Enzymes associated with GAG catabolism are lysosomal hydrolases • Patients with MPS have less than 1%residual enzyme activity • GAGs accumulate in lysosomes – this results in cellular dysfunction and clinical abnormality
MPS Classification
MPS Classification • Four broad categories based on dominant clinical features o Soft tissue storage and skeletal disease with or without brain disease (MPS I, II, VII) o Soft tissue and skeletal disease(MPS VI) o Primary skeletal disorders (MPS IVA , IVB) o Primary central nervous system disorders ( MPS III A-D)
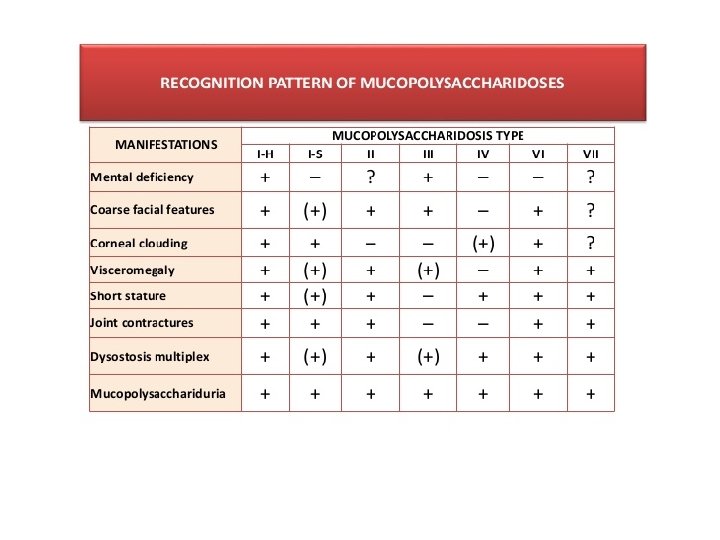
Clinical presentation • Many physical disorders • Varying degree of severity depending on MPS type • Features may not be apparent at birth but progress as storage of GAGs increase
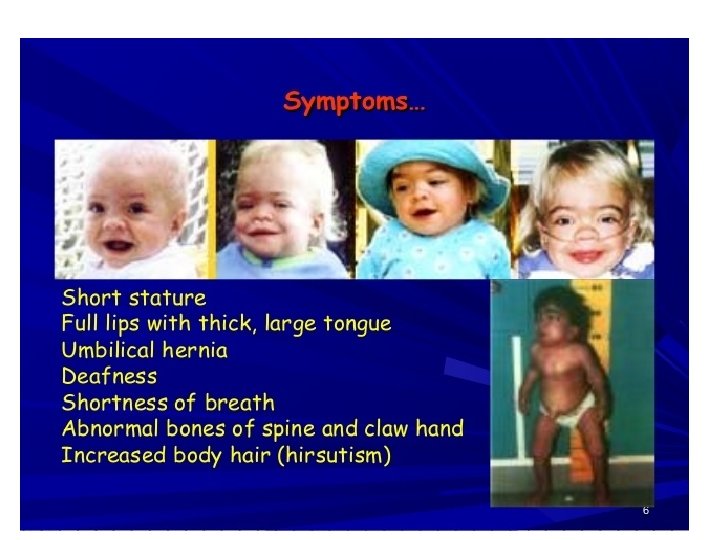
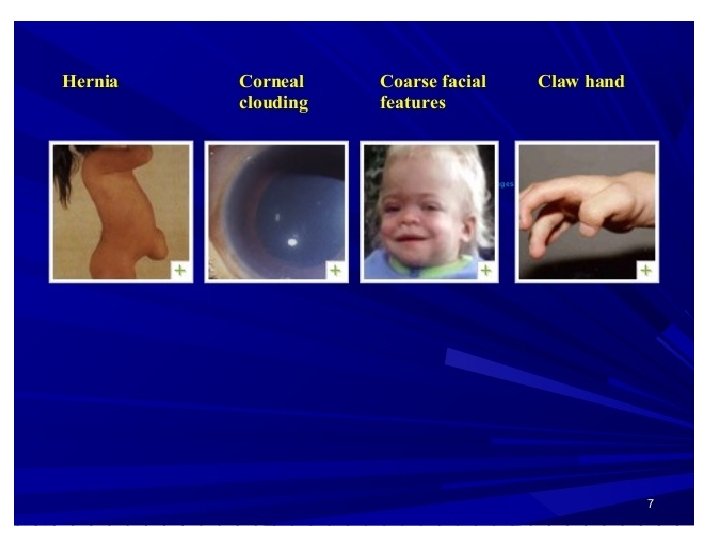
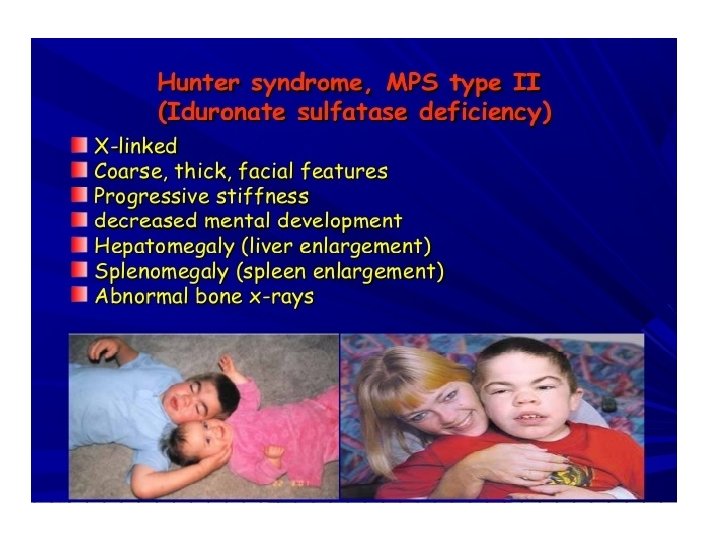
Common presentations • CNS disease – mental retardation , developmental delay , severe behavioral problems , hydrocephalus • CVS disease- Valvular dysfunction • Pulmonary disease- airway obstruction, sleep apnoea • Opthalmologic – corneal clouding • Hearing impairment – deafness • Musculoskeletal disease- short stature, joint stiffness • Others – coarse facial features , hepatosplenomegally , hernia
Hurler , Hurler-Scheie, Scheie syndrome(MPS I) • • Autosomal recessive mode of inheritance Due to lysosomal α-L- Idoronidase deficiency Newborns are normal at birth Clinical features start manifesting by age 1 year
Clinical features • • • Developmental delay Coarse thick facial features Low nasal bridge Prominent dark eye brows Progressive joint stiffness Severe mental retardation
Diagnosis • High index of suspicion- a child with coarse facial features, bone disease, developmental delay, short stature, hepatosplenomegally , corneal clouding • GAG urinary concentration • Lysosomal Enzyme assay – definitive diagnosis from cultured fibroblasts, leukocytes • Prenatal diagnosis in selected family clustersamniocytes and chorionic villus culture
Management of MPS • Goal- to reduce severity of symptoms and improve quality of life • Multidisciplinary team care v. Neurologist v. Pediatrician v. Cardiologist v. Ophthalmologist v. Audiologist v. Orthopedic surgeon v. Physical and occupational therapist
Management • Supportive care especially physical therapy is critical • Main treatment modalities are v. Haematopoietic stem cell (peripheral blood leukocytes) transplant (HSCT) for MPS I and II. Has high risk of morbidity and mortality from cardiac and pulmonary complications. Reduces clinical progression of some features in some children v. Enzyme replacement therapy (ERT)- MPS I, II, VI • Treatment results in improvement of somatic disease
Thank you Asanteni
Reference • Joseph Muenzer. Overview of the mucopolysaccharidosis, , Rheumatology, Vol 50, 2011 • Thomas J et al. Diagnosis of mucopolysaccharidosis, Rheumatology , Vol. 50, 2011 • Lone Clark. Mucopolysaccharidosis type 1. Gene reviews 2016