Biochemie neurodegenerativnch chorob Alice Skoumalov Neurodegenerativn choroba Patologick

Biochemie neurodegenerativních chorob Alice Skoumalová
 Alzheimerova choroba β-amyloid Parkinsonova choroba α-synuclein Frontotemporální demence Tau")
Neurodegenerativní choroba Patologický protein (amyloid) Alzheimerova choroba β-amyloid Parkinsonova choroba α-synuclein Frontotemporální demence Tau Huntingtonova choroba Huntingtin Creutzfeldt-Jakobova nemoc Prion
 specifických proteinů β-skládaný list")
Nemoci způsobené patologickou konformací proteinů Způsobené abnormální terciální strukturou (konformací) specifických proteinů β-skládaný list α-helix konformační změna Normální protein (nativní struktura) Protein způsobující onemocnění (patologická konformace) Agregace Toxická aktivita Neurodegenerativní choroby Ztráta funkce

DNA Ubikvitin RNA Ribosom ATP Chaperony Nativní protein Chaperony Protein s patologickou konformací Agregát/fibrilární amyloid Proteasom Ukládání (Amyloidosy) Degradovaný protein Toxická bílkovina (Alzheimerova choroba) Ztráta funkce (Cystická fibrosa)

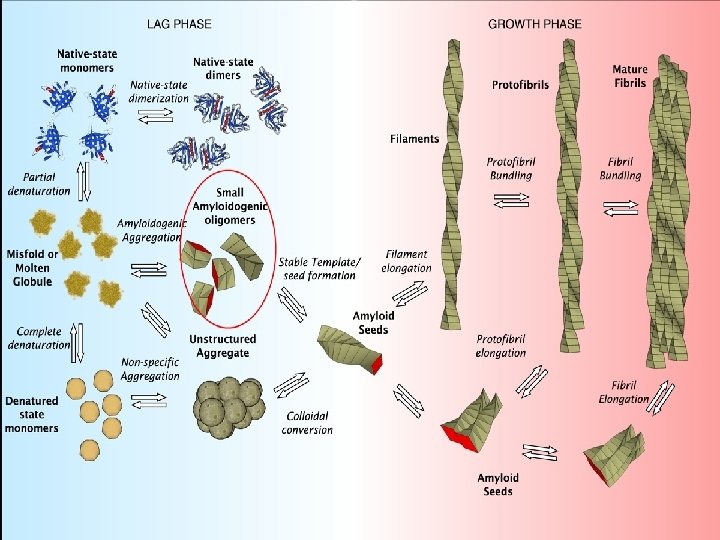
Struktura amyloidních fibril Ø Rovné, nerozvětvené, velikost 8 -16 nm Ø Složeny ze 2 až 6 protofilament (3 -4 nm) Ø Bohaté na struktury β-struktury (vzájemně spojeny vodíkovými můstky)

Molekulární faktory tvorby amyloidu Ø Abnormální zavinutí proteinu je klíčové pro tvorbu amyloidu Ø Stabilita proteinu (stálost nativní konformace proti abnormální) je důležitý faktor pro náchylnost k tvorbě amyloidu Destabilizační faktory: 1. Extrémní podmínky prostředí (např. kyselé buněčné kompartmenty) 2. Proteolytické štěpení proteinů (endogenní proteasy) 3. Mutace, které pozmění primární strukturu (mnoho amyloidos má familiární formu způsobenou mutací proteinovém prekursoru amyloidu) 4. Kontakt s lipidovou dvojvrstvou membrán (Aβ)

Kontrola kvality proteinů

Biochemie Alzheimerovy choroby

Prevalence ACH v populaci Ø Roste s věkem Ø 50% po 85. roce

Typy ACH Familiární Sporadická Ø < 2%, AD Ø Nástup před 65. rokem Ø Mutace : Ø Ø § APP (chromozom 21) § PSEN 1 (chromozom 4) § PSEN 2 (chromozom 1) Většina Nástup později Neznámé příčiny Genetický faktor: § polymorfismus Apo. E
 plaky Ø hyperfosforylovaný tau")
Základní patologické znaky provázející Alzheimerovu chorobu Neurofibrilární tangly Amyloidní (senilní) plaky Ø hyperfosforylovaný tau protein Ø fibrily proteinu β-amyloidu (Aβ) Øabnormální zpracování APP (amyloid prekurzor protein)

Úloha Aβ v patogenezi ACH Štěpení APP a vznik Aβ 1. Neamyloidogenní kaskáda (α-sekretasa, γ-sekretasa) 2. Amyloidogenní kaskáda (β-sekretasa, γ-sekretasa) Úloha lipidových raftů ! Upraveno podle: Thomas, Fenech. Mutagenesis 2007 22: 15 -33.

Enzymový komplex γ-sekretázy

Tvorba iontových kanálů v membráně

Hlavní faktory patogeneze ACH Upraveno podle: Jellinger et al. J Cell Mol Med 2008, 12: 1094 -1117

Amyloidní kaskáda - patologie Alzheimerovy choroby Mutace APP, PS 1 a PS 2 genů Zvýšená produkce a kumulace Aβ 42 Oligomerizace Aβ 42 a depozice v senilních placích Ovlivnění synapsí oligomery Aβ 42 Aktivace mikroglií a astrocytů (komplement faktory, cytokiny) Progresivní poškození synapsí a neuritů Poškození iontové rovnováhy neuronů, oxidační poškození Ovlivnění signálních drah kináz a fosfatáz tvorba tanglů Poškození funkce neuronů a buněčná smrt s deficitem transmiterů Demence

Úloha zánětu v patogenezi ACH Upraveno podle: Block. BMC Neuroscience 2008, 9: 1 -8.

Úloha oxidačního stresu v patogenezi ACH
 • Problematika sestřihu APP")
ACH shrnutí • Klíčovým faktorem patogeneze je Aβ (oligomery, plaky) • Problematika sestřihu APP (deposita amyloidu) • Rozvoj zánětu v mozku • Přítomnost oxidačního stresu

Biochemie prionových chorob

Definice prionů Ø proteinové přenosné patogeny způsobující některé fatální neurodegenerativní choroby (např. u lidí Creutzfeld. Jakobovu nemoc, kuru, u zvířat bovinní spongioformní encefalopatii) Ø zkratka proteinaceous infectious particle (analogie virion); jde o infekční agens, které nenese genetickou informaci uloženou v nukleové kyselině! Ø proteiny s patologickou konformací, které jsou schopny dále propagovat konformační změny nativních proteinů na abnormální struktury

Nemoc Hostitel Pr. P isoforma Scrapie Ovce, koza Ov. Pr. PSc Transmissible mink encephalopathy (TME) Norek Mk. Pr. PSc Chronic wasting disease (CWD) Jelen, los MDe. Pr. PSc Bovine spongioform encephalopathy (BSE) Skot Bov. Pr. PSc Feline spongioform encephalopathy (FSE) Kočky Fe. Pr. PSc Exotic unguale encephalopathy (EUE) Antilopa Nya. Pr. PSc Kuru Člověk Hu. Pr. PSc Creutzfeldt-Jakob disease (CJD) Člověk Hu. Pr. PSc Gerstmann-Straussler. Scheinker syndrome (GSS) Člověk Hu. Pr. PSc Fatal familial insomnia (FFI) Člověk Hu. Pr. PSc
 Název Pr. PSc (scrapie) Normální protein")
Pr. PC Pr. PSc Název Pr. PC (cellular) Název Pr. PSc (scrapie) Normální protein Abnormální protein způsobující nemoc Transmembránový glykoprotein (neurony, lymfocyty); funkce není známá; váže Cu 2+ (homeostasa těchto iontů) Stejná sekvence aminokyselin (primární struktura) Dominantní sekundární struktura α-helix Dobře rozpustný ve vodě Monomerní, snadno štěpen proteasami Kódován genem zvaným PRNP na chromosomu 20 Dominantní sekundární struktura βskládaný list Nerozpustný ve vodě Multimerní, rezistentní k působení proteas Když je v kontaktu s normálním proteinem Pr. PC, konvertuje ho na abnormální Pr. PSc Tvoří agregáty (způsobují onemocnění a/nebo vznikají jako vedlejší efekt)
 Pr. PSc β-skládaný list (40%),")
Molekulární model struktury: Pr. PC Převažuje α-helix (3 x) Pr. PSc β-skládaný list (40%), α-helix (30%)
")
Prionové agregáty (snímek z elektronového mikroskopu)
 nebo endogenní (vrozená či sporadická forma)")
Replikační cyklus ØPřítomnost iniciálního Pr. PSc: exogenní (infekce) nebo endogenní (vrozená či sporadická forma) ØAbnormální protein Pr. PSc konverguje nativní protein Pr. PC na Pr. PSc v replikačním cyklu ØPatologické Pr. PSc agregují

Mechanismus progrese prionů
 Ø Ø progresivní choroby, atakují mozek a")
Prionové choroby: vzácné neurodegeneratvní onemocnění (1 nemocný/milion) Ø Ø progresivní choroby, atakují mozek a nervový systém, potenciálně přenosné priony Klinika: ztráta motorických funkcí (poruchy koordinace, ataxie, nechtěné trhavé pohyby), změny osobnosti, deprese, nespavost, zmatenost, poruchy paměti, demence, progresivní svalová paralysa, smrt Definitivní diagnosa: post mortem histologicky Neléčitelné 1. Sporadické (85 %) U starších lidí, rychlá progrese (1 rok) Př. Creutzfeldt-Jakob disease (CJD) 2. Familiární (vrozené-15%) Mutace Pr. P genu způsobí změnu nativní konformace na patologickou Pr. PSc Př. Gerstmann-Straussler-Scheinkerova nemoc (GSS), fatální familiární insomnie (FFI) 3. Přenosné (velice vzácné, ale mediálně známé) V laboratoři přenosné na myši intrakraniální injekcí mozkového homogenátu nakažených zvířat Přenos nemoci kuru u lidí (domorodci na Nové Guinei-kanibalismus) Nedávno zjištěn přenos BSE na člověka v Evropě po požití infekčního hovězího masa (varianta CJD)

Přenos prionů 1. 2. Přímý kontakt s infikovanou tkání: např. iatrogenní přenos CJD u pacientů, kterým byl aplikován růstový hormon získávaný z lidské hypofýzy, z instrumentů u mozkové chirurgie (priony přežijí běžnou sterilizaci), rohovkové transplantáty, elektrodové implantáty Konsumpce infekčního masa: např. byl prokázán přenos prionů u nemoci kuru (kanibalismus u domorodců z Nové Guinei) a v. CJD (požití masa z krav s BSE) Není znám mechanismus jak se priony dostanou z trávicího traktu do mozku (normálně jsou proteiny tráveny a vstřebávány jako aminokyseliny) Hypotesa: Nejsou tráveny (rezistence k proteasam), jsou transportovány přes lymfatickou tkáň střeva (gut-associated lymphoid tissue GALT)
 Nejčastější prionové onemocnění Výskyt mezi 55 -65 roky (varianta CJD u")
Creutzfeldt-Jakobova choroba (CJD) Nejčastější prionové onemocnění Výskyt mezi 55 -65 roky (varianta CJD u mladších) Do 1 roku smrt Klinika: Demence, halucinace, poruchy motoriky Diagnosa: Klinické symptomy, EEG, MRI, analysa CSF (nárůst 14 -3 -3 proteinu) Definitivní diagnosa: histologie mozkové tkáně Léčba: neléčitelná, hledání léků Formy: 1. Sporadická 2. Familiární 3. Přenosná: iatrogenně-i. CJD, požitím-v. CJD Dárcovství krve: přenos transfuzí, nedá se zjistit u dárců, restrikce pro dárce
 Prionové onemocnění vyskytující se mezi domorodci")
Kuru (domorodé slovo znamenající „roztřesený zimou a horečkou“) Prionové onemocnění vyskytující se mezi domorodci na Nové Guinei (poprvé diagnostikováno 1900) 1950 -60 epidemie Kanibalismus: příbuzní pojídali mozky mrtvých na znamení smutku V 50. letech zákaz kanibalismu kvůli přenosu TSE, koncem 20. století nárůst nemocných (inkubační doba 30 -50 let) Symptomy: 3 fáze, postupně se zhoršující motorické a mentální funkce

Typy prionů =subklasifikace prionů na základě strukturálních charakteristik, které definují jejich patologický profil Liší se inkubační dobou a neuropatologickými znaky, i když jsou způsobeny stejným proteinem (Pr. PSc) Existence různých typů prionů: jsou chemicky identické, ale nemají přesně stejnou konformaci Strukturální charakteristiky jednotlivých prionových typů jsou propagovány přes replikační cyklus Jednotlivé typy prionů mají specifické cílové tkáně, inkubační dobu a patogenezi

Různá inkubační doba a neuropatologie typů prionů -různé biochemické charakteristiky -odlišnosti jsou zachovány po opakovaném přenosu Charakteristiky jednotlivých typů prionů jsou zachovány po přenosu na jiné druhy -po re-izolaci v původním hostitely

Konsumpce BSE-nakaženého masa Prion BSE Varianta CJD: u mladších, jiný klinický průběh Prion v. CJD je identický s prionem BSE Klasická CJD: u starších Prion CJD se liší

Charakteristika Klasická CJD Varianta CJD Věk úmrtí 68 let 28 let Doba trvání nemoci 4 -5 měsíců 13 -14 měsíců Klinické symptomy Demence, časné neurologické projevy Psychiatrické projevy, pozdější neurologické poruchy, senzorické halucinace Specifický nález na MRI Přítomen Specifický nález na EEG Přítomen Nepřítomen Imunohistochemie mozkové tkáně Variabilní akumulace Pr. PSc Významná akumulace Pr. PSc Nález prionů v lymfatické Nesnadno detekovaný tkáni Snadno detekovaný Přítomnost amyloidních plaků v mozku Přítomny

Konformační selekce a druhová bariéra Ø Široká škála možných konformací Pr. PSc Ø Pro jednotlivé druhy termodynamicky možné pouze některé z nich Ø Stejné konformace - možný přenos

Terapeutické strategie 1. Léky, které přeruší replikační cyklus Pr. PSc Takové léky byly testovány na buněčných modelech, musí být ověřeny na zvířecích modelech a testovány v klinických studiích 2. Vývoj vakcíny: abnormální protein, na rozdíl od nativní formy, exponuje specifické aminokyselinové zbytky; protilátky proti této specifické sekvenci stimulují imunitní reakci proti patologickému prionu 3. Vývoj peptidů rozrušující struktury β-skládaného listu 4. Genová terapie: úprava genu kódujícího priony Genetické inženýrství: kráva s chybějícím genem produkci prionů; teoreticky imunní k BSE (prosinec 2006)

Priony shrnutí Ø Priony jsou proteiny, které nesou informaci pro vlastní reprodukci (vyvrácení dogmatu moderní biologie) Ø Priony se nacházejí v membránách u zdravých lidí a zvířat, ale jejich abnormální konformace (Pr. PSc) jsou nerozpustné, nedají se štěpit a agregují Ø Pr. PSc atakuje nativní prion Pr. PC, změní jeho konformaci na abnormální, způsobí exponenciální nárůst nerozpustných proteinů, které agregují a tvoří fibrilární struktury Ø Prionové choroby jsou vzácné neléčitelné degenerativní poruchy, některé z nich jsou přenosné, ale mechanismus není úplně jasný (např. přenos BSE na člověka)
 Ø Oligomerní agregáty")
Závěr Ø Konformační defekt proteinů - amyloid - PCD (neurodegenerativní nemoci) Ø Oligomerní agregáty patologických proteinů neurotoxické Ø Zánět a oxidační stres Ø Terapeutické strategie
- Slides: 40