Selected syndromes every neurologist should know Myriam Srour
































 – – –")







- Slides: 42
Selected syndromes every neurologist should know Myriam Srour, Pediatric Neurologist September 25, 2013
Retts syndrome
Classical Retts- main clinical features 1. 2. 3. 4. Girls Initial normal development followed by regression (6 -18 mo) Deceleration of head growth Loss of purposeful hand movements
1. 2. 3. 4. Main criteria Partial or complete loss of acquired purposeful hand skills Partial or complete loss of acquired language skill Gait abnormalities (dyspraxic or absence of ability) Stereotypic hand movements including handwriting/squeezing, clapping/tapping, mouthing, and washing/rubbing automatisms Supportive criteria • • • Breathing disturbances when awake Bruxism when awake Impaired sleep pattern Abnormal muscle tone Peripheral vasomotor disturbances Scoliosis/kyphosis Growth retardation Small cold hands and feet Inappropriate laughing/screaming spells Diminished response to pain Intense eye communication- “eye pointing”
4 Stages
Other features • Epilepsy – In 50 -90% – All types – Onset usually between 2 -5 years – Seizure frequency declines with age (late childhood/adolescence) • Non-epileptic events – In one study, only 1/3 of reported seizures were correlated with abnormal activity on EEG – Hand stereotopies, breath-holding, hyperventilation, dystonia, dyskinesias, unusual eye movements • Bowel dysmotility • Gallbladder dysfunction, stones • Osteopenia • Prolonged QT interval – Avoid medications that prolong QT
Different forms • Classic Retts- typical Retts – Presence of all 4 major criteria • Atypical Rett syndrome – Period of regression – 2 of 4 main criteria – 5 of 11 supportive criteria
Retts syndrome variants 1. Epileptic encephalopathy variant – Seizures predominate – Onset prior to 6 months – Think of CDKL 5 mutations 2. Congenital variant – No regression – Think of FOXG 1 mutations 3. Later onset variant 4. Forme fruste variant 5. Preserved speech variant
Genetics • Mutations in MECP 2 – X-linked – Lethal in males • Neonatal encephalopathy in boys, severe, death <2 years – Mutations are de-novo – Methyl-Cp. G-binding protein 2 – Abundantly expressed nuclear protein – Mediates transcriptional silencing and epigenetic regulation of methylated DNA
Key concept Methylation usually mediates silencing MECP 2 recruited to methylated sites This leads to recruitment of proteins that will de-acetylate histone tails Chromatin compaction (inactive genes)
What are the main features?
Main clinical features of Fragile X? • Dysmorphisms – More obvious in adults/post-pubertal • • • Long face Large ears Prominent chin Macroorchidism Hyperextensible joints strabismus
Main clinical features of Fragile X? • Intellectual disability – Moderate to severe (IQ 30 -50) – Variable • ASD features – Impulse control problems, poor eye contact, perseverative speech – 20 -80% of males, 10— 20% females • Cardiac • Mitral valve prolapse • Aortic root dilatation
Fragile X genetics • X-linked, Dominant • Triplet repeat disorder • FMR 1 testing – Testing triplet repeats CGG in 5’UTR – PCR/Southern blot – Why is it called fragile X? • Fragile site on X chromosome when cells grown in folate-deficient medium • Triplet repeats decrease transcription of FMR 1 (ie. Less m. RNA) • What does FMRP do? – Found in the cytoplasm of most cells, especially neurons – FMRP binds RNA and functions as a nucleocytoplasmic shuttling protein – Plays an important role in the structural and functional maturation of synapses by suppressing translation of certain genes. – Disrupts glutamatergic neurotransmission.
Fragile X- genetics • What is a pre-mutation? – 50 -200 repeats • How many repeats are in a full mutation? – >200 • What happens to the repeats with future progeny – In male carrier? – Female carrier? • Increase in number of repeats if transmitted by female, but stable if transmitted by male
Fragile X • How prevalent is Fragile X amongst patients with ID/ASD? – 1. 5 -3% • What do you counsel a male with Fragile X premutation? – Risks to children – Risks to grandchildren – Risks to self – FXTAS
FXTAS • Fragile X-related tremor/ataxia syndrome – Males with pre-mutation – Onset after 50 (usually early 60 s) – Intension tremor and or/gait ataxia – Mild parkinsonism – Neuropathy – MRI: • T 2/FLAIR lesions in Middle cerebellar peduncle • Also in periventricular white matter and brainstem • Atrophy
FXTAS • With pre-mutation, there is increased transcription of FMR 1 (i. e. more m. RNA). • Pathogenesis results from neural toxicity of FMR 1 m. RNA • Inclusion in neurons and astrocyes, spongiform white matter changes in subcortical, periventricular and brainstem regions including middle cerebellar peduncle.
FXTAS • What do you counsel a female with Fragile X premutation? – To children – To self – Premature Ovarian failure in 30%
Angelman Syndrome • Protruding tongue • Widely spaced teeth • Happy/smiling • Uplifted, flexed armed position • Prognathia
Main clinical features • GDD/ Intellectual disability – Severe range, best have 20 words – No regression • Happy demeanor – “happy puppet” – Laughing, hand flapping • Ataxic gait/tremulousness • Microcephaly • Seizures – 90%, usually start <3 years • Sleep disturbances • Fascination with water
Characteristic EEG pattern - High amplitude sharp theta posteriorly
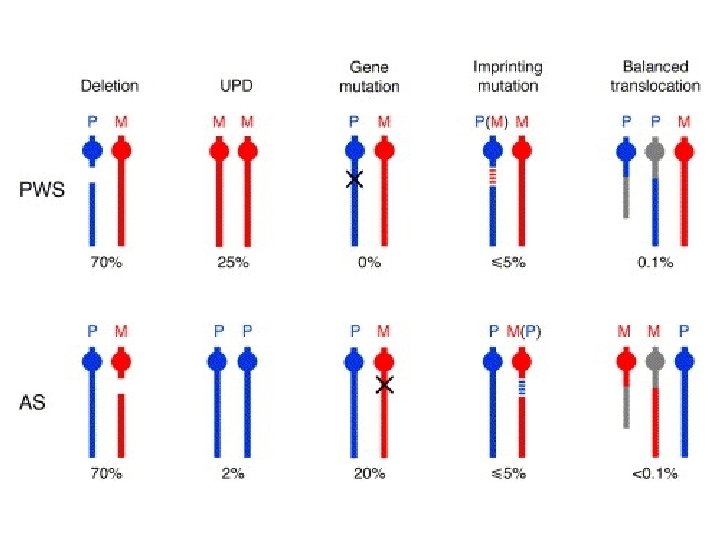
Genetics- key concept What is genomic imprinting? - Genes are expressed in a parent-of origin manner • i. e. expression depends on whether it is inherited from the mother or father • e. g. deletion of 15 q 11 if maternal Angelman syndrome if paternal Prader-Willi syndrome
Genetics • due to deficient expression or function of maternally-inherited UBE 3 A • What are 4 genetic mechanisms underlying Angelman syndrome? 1. 2. 3. 4. Deletion 15 q 11 (65 -70%) Paternal uniparental disomy (2 -5%) Imprinting defect (2 -5%) UBE 3 A point mutation (10 -15%) Pick up on methylation studies
What is uniparental disomy? Inheritance of 2 copies of a chromosome/part of chromosome from the same parent and no copies from the other parent
Genetic mechanisms
What is function of UBE 3 A? • Ubiquitin-protein ligase E 3 A – Involved in the ubiquitination pathway, which targets slected protein for degradation
PWS- other clinical features Hypothalamic dysfunction • Lack of satiety – Low ghrelin levels – Hypogonadism in males and females • Hypothyroidism • Adrenal insufficiency • Hypogonadism, infertility
PWS- Clinical features • Characteristic facial features: almond-shaped eyes, blond, thin upper-lip, down-turned mouth • Type-2 diabetes + complications of obesity • Behavior issues: temper tantrums, stubborn, compulsive, autistic features, psychotic features
Prader-Willi syndrome • Main Clinical features • Age dependent! Birth – 2 y Hypotonia with poor suck 2 -6 y Global developmental delay, hyperphagia begins Hyperphagia, behaviour abnormalities 6 -12 y Teensadult Mild MR, hypothalmalic hypogonadism
PWS- genetics • Absence of Paternal 15 q 11 contribution (PWCR) – – – Deletion- 70% Maternal uniparental disomy- 20 -30% Imprinting defect- Mutation in methylation center Same region as Angelman syndrome Methylation testing identifies 99%
Down syndrome
Down syndrome • • • Upslanting palpebral fissures Epicanthal folds Flat profile Folded, dysplastic ears Low-set ears Brachycephaly Open mouth, protruding tongue Excessive skin at nape Transverse palmar crease Incurved 5 th finger Sandal gap Hypotonia and joint hyperlaxity
Trisomy 21 • 88% due to maternal non-dysjunction • 1 -2% individuals mosaic Down’s • 2 -3% due to Robertsonian (balanced translocation) – Recurrence risk implications
Screening • 1 st trimester combined test: – Ultrasound for nuchal translucently – Serum markers (beta-h. CG and PAPP-A) – 85% sensitive, 5% false positive • Chorionic Villus sampling • > 35 years high-risk • Offered to all women
Clinical features • GDD/Intellectual disability – Variable. Mainly mild-moderate • • Cardiac defects (VSD, CAVSD) Vision, hearing problems Short stature GI (duodenal atresia) Endocrine disorders Hematologic disorders (leukemias) Atlantoaxial instability Immune deficiencies
Neurologic features • Hypotonia • Ligamentous laxity – Atlanto-axial instability – Signs and symptoms consistent with spinal injury • Seizures – Bimodal distribution: infancy or after 3 rd decade – Infantile spasms (usually more easily controlled) – Seizures >30 years associated with dementia • Dementia – Alzheimers- typical neuropathology with senile plaques and tangles – Related to increased dosage of APP on chromosome 21 (amyloid precursor protein) • Obstructive sleep apnea
QUESTIONS?