Mi a termodinamika Alapfelttelezse a korpuszkulris elmlet a












 Egykomponensű rendszerek F Sz 1")
 Kétkomponensű rendszerek F 1 Sz")

 1737 -ben tervezte meg a ma is általánosan használt (100")

 körülmények")
 p Vm = R")
 Egysége: Pascal (N/m 2) 1 bar = 105 Pa")
 Parciális nyomás: pi")

 mérése • anyagmennyiség (mól)")
 V = állandó")
 (Boyle–Mariotte) p. V = állandó [ha")
")
 T Gay-Lussac I. izobar p = (n R /V)")
 p")
 = 0")
 A gázok korlátlanul elegyednek egymással. A tökéletes gázok ideális elegyet alkotnak.")


 között nem érvényesek.")








 kapcsolata: a kritikus")






 Térfogati munkában mindig a külső nyomás (p. K) szerepel. reverzibilis változás: p")



 Melegítés-hűtés: Q = c · m · T c = fajlagos hőkapacitás (fajhő)")

 Fázisátalakulás A fázisátmenetek izoterm és izobár folyamatok. Tiszta anyag esetén vagy a hőmérsékletet")
, a belsőenergia-változás a hővel")

 az entalpiaváltozás a hővel egyenlő. Az entalpia-változás számítása")
: - moláris párolgáshő Hm (olvadás): - moláris olvadáshő Az entalpia a")




 mert állandó nyomáson végzett melegítéskor kiterjed a")
")
 A reverzibilis állapotváltozásokat tárgyaljuk. A gázok valóságos folyamatai")
: Belsőenergia-változás:")
 Térfogati munka: W=0 Hő (belsőenergia-változás): Entalpia-változás:")



:")

 V és T kapcsolata Itt vezetjük be azt a")
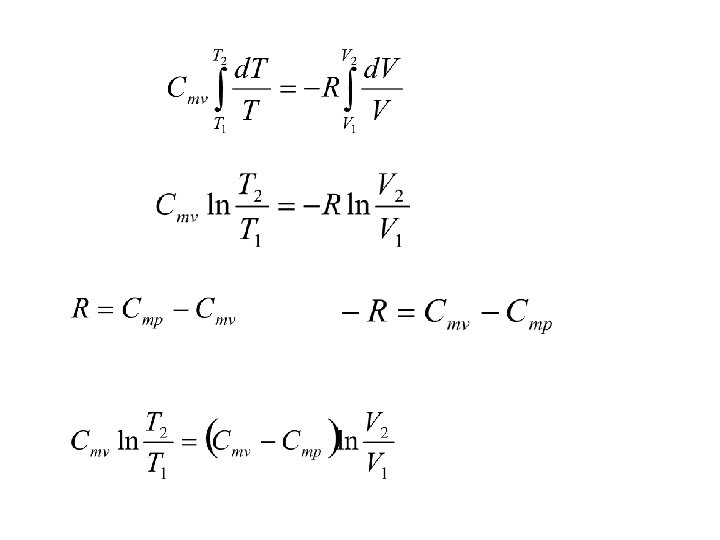
")

 p és T kapcsolata")

 és endoterm (hőemésztő) reakciók")




 következik, hogy a standard reakcióhő lényegében")



: q = C· T, C a kaloriméter")

 +7, 5 O 2 = 7 CO 2(g)")

 + O 2 = CO 2 (1) reakció entalpia -változása megegyezik")



 végbemenő képződési")
, majd")


 és po = 105 Pa nyomáson az")






 rendszer olyan nyitott rendszer, amelyben az állapotfüggvények függnek a helytől, de")


- Slides: 112
Mi a termodinamika? Alapfeltételezése a korpuszkuláris elmélet: a rendszerek szabad szemmel nem látható elemi részekből tevődnek össze, amelyekre első közelítésben érvényesek pl. a mechanika törvényei. A részecskékre a mechanikai differenciálegyenleteket felírva egy igen sok ismeretlent (részecskék helye, sebessége) tartalmazó egyenletrendszer írja le a rendszer viselkedését. Miután ez mind gyakorlatilag, mind elméletileg (Heisenberg) megoldhatatlan, a termodinamika a mikroszkopikus mennyiségek helyett makroszkopikus, statisztikus (átlagos) mennyiségekkel dolgozik (pl. nyomás, belső energia, hőmérséklet). Ezek között az un. fundamentális egyenlet teremt kapcsolatot.
Mi a termodinamika? A termodinamika további jellemzője, hogy a mechanikától, stb. eltérően itt a folyamatok egyirányúak, nem megfordíthatók (a veszteségek miatt, folyamatoknál entrópia-produkció). Az intenzívek (hőmérséklet, nyomás, . . . ) jellemzik az kialakuló extenzíváramok (belső energia, tömeg, . . . ) irányát. Így ez az első olyan leírás, amelyik egy fizikai rendszer evolúciójával (fejlődés, haladás, természettudományos materializmus, darwinizmus) számol. A hagyományos TD elsősorban energetikailag írja le egy fizikai rendszer viselkedését, az energiatétel használatával ugyanis sok feladat egyszerűbben megoldható, mint más módszerekkel. Ugyanakkor termodinamikai módszerekkel (statisztikus extenzív és intenzív mennyiségek, fund. állapotegyenlet, entrópia) dolgoznak más tudományágak is (pl. közgazdaságtan).
Homogén: makroszkopikus tulajdonságok minden pontban azonosak. Inhomogén: egyes makroszkopikus tulajdonságok helyről helyre változnak; eloszlásukat folytonos függvény írja le. Heterogén: ugrásszerűen változó makroszkopikus tulajdonságok. Pl. jég-víz rendszer Fázis: a rendszer homogén kémiai összetételű és homogén vagy inhomogén fizikai szerkezetű része. A fázis lehet diszpergált (széttöredezett), ilyenkor egy fázisba soroljuk az azonos összetételű részeket. Komponens: a rendszernek a kémiai tulajdonság alapján megkülönböztethető része.
A termodinamikai rendszer állapota a mérhető fizikai tulajdonságok összessége. A rendszer állapotától függő makroszkopikus jellemzőket állapotjelzőknek (állapothatározóknak) nevezzük. Az alap-állapot jelzők: tömeg (anyagmennyiség) m (n) térfogat V nyomás (p) hőmérséklet (T) koncentráció (c)
A rendszer termodinamikai egyensúlyban van, ha az állapothatározók egyike sem változik. Egyensúlyban nem játszódnak le makroszkopikus folyamatok. Nem egyensúlyi rendszer: állapothatározók az időben változnak. Reverzibilis változás: végállapotból ugyanazon közbülső egyensúlyi állapotokon keresztül jut a rendszer a kezdeti állapotba. Olyan folyamat, amelyet a változók infinitezimális módosításával meg lehet fordítani. A valóságos folyamatok mindig irreverzibilisek.
Pl. egy gáz reverzibilis összenyomása azt jelenti, hogy a külső nyomás csak észrevétlenül nagyobb, mint a gáz nyomása, tehát a rendszer és környezete között mechanikai egyensúly van. A valóságos folyamatok sok esetben jól megközelítik a reverzibilis határesetet. Gyakran vizsgált folyamatok: Izoterm ( t=áll. ) izobár (p =áll. ) izosztér, izochor (V = áll. ) adiabatikus (Q = 0)
Az állapotfüggvény az állapothatározók olyan többváltozós egyértékű függvénye, amelynek változása csak a kezdeti és végállapottól függ. Független az úttól, amelyen a rendszer a kezdeti állapotból a végállapotba jutott. (pl. potenciális energia a gravitációs térben). Legfontosabb állapotfüggvények: U – belső energia H – entalpia S – entrópia A – szabadenergia G – szabadentalpia Változás pl. U Infinitezimális változás: d. U (teljes differenciál).
Útfüggvények: értékük függ a kezdeti és végállapot között megtett úttól. Ilyen pl. a munka és a hő. Pl. vízszintes súrlódó felületen A pontból eljuttatunk egy tárgyat a B pontba W 2 W 1 Változásról nem beszélünk. Infinitezimális érték: W, Q nem teljes differenciál, mert az integrálásához további adatokat kell megadni.
A termodinamikában használt mennyiségeket egy más szempontból is osztályozhatjuk: Extenzív mennyiségek: függnek a rendszer kiterjedésétől és additívak : tömeg (m) térfogat (V) belső energia (U), stb. Intenzív mennyiségek: nem függenek a rendszer méretétől, és nem additivak: hőmérséklet (T) nyomás (p) koncentráció ( c)
Extenzív mennyiségből csinálhatunk intenzív mennyiséget, ha azt egységnyi tömegre, térfogatra, stb. vonatkoztatjuk. Sűrűség = m/V Móltérfogat: Vm = V/n Moláris belső energia: Um = U/n Állapotegyenlet: az egyensúlyban levő rendszer állapotfüggvényei között teremt kapcsolatot. Pl. tökéletes gáz állapotegyenlete: p. V = n. RT R = 8, 314 Jmol – 1 K-1 V m 3 T K p Pa n mol Valóságos anyagok állapotegyenletei empirikus függvények ( hatványsor, diagram, táblázat formájában).
A fázisok és komponensek számának kapcsolata Fázisok száma: F Komponensek száma: K Szabadsági fokok száma: SZ: azon intenzív állapotjelzők száma, amelyek bizonyos határon belül szabadon változtathatók, anélkül, hogy a fázisok száma megváltozna. SZ = állapotjelzők száma - egyenletek száma
Minden egyes fázisban K-1 adattal jellemezhetjük a koncent-rációkat. Pl. metán-propán elegy. Ha tudjuk az első kettő móltörtjét, a harmadik kiszámítható: ypr = 1 - (ym +ye). Az állapotjelzők száma tehát minden fázisban K+1 (K-1 móltört adat + a nyomás és a hőmérséklet). F számú fázisban F(K+1) adat Egyenletek száma: minden intenzív mennyiségre F-1 egyenlet (pl. p 1 = p 2 = …= p. F). Koncentrációkra: megoszlási egyensúly. K+2 intenzív állapotjelző van. Összesen (K+2)(F-1) egyenlet Sz = F(K+1) - (K+2)(F-1) = K-F+2
Sz = K - F + 2 (Gibbs-féle fázisszabály) Egykomponensű rendszerek F Sz 1 2 (T, p) 2 1 3 p 0 (hármaspont) szilárd folyadék fluid gáz O T
Sz = K - F + 2 (Gibbs-féle fázisszabály) Kétkomponensű rendszerek F 1 Sz p 3 (T, p, x) 2 2 3 1 Síkban csak úgy ábrázolhatjuk, ha az egyik állapotjelzőt rögzítjük. t = áll. folyadék P 1* 0 P 2* gőz x, y 1
A termodinamikai hőmérséklet és nyomás A hőmérséklet fogalma a hideg – melegérzetből fejlődött ki. A ma legelterjedtebb hőmérsékletskálát 1742 -ben javasolta a svéd Andres Celsius.
Andres Celsius (1701 -1744) 1737 -ben tervezte meg a ma is általánosan használt (100 fokos beosztású) hőmérsékletskálát, melynek azóta is megorízte nevét, sőt az egyik leggyakrabban elhangzó névvé tette világszerte. Ötlete, amelyet 1742 -ben ismertetett a Svéd Akadémián tartott előadásában, leegyszerűsítette a hőmérsékletmérést, és a kapcsolódó számításokat. Celsius azonban a forráspontot jelölte 0 -val, s a fagyáspontot 100 -al, a két számot 1750 -ben Stromer svéd tudós cserélte fel.
A Celsius-skála két alappontja: olvadó jég: 0 C forrásban lévő víz: 100 C Milyen anyag milyen fizikai tulajdonságát használjuk a hőmérséklet mérésére? Folyadékok (pl. higany vagy alkohol) hőtágulálása. Nem használhatók széles hőmérsékleti tartományban. Ugyanazt a hőmérőt más folyadékkal töltve nem ugyanazt az értéket mutatja, mert a folyadékok hőtágulása különbözik (szigorúan véve nem arányos). Pl. Hg-nyal : 28, 7 C-ot, alkohollal 28, 8 C-ot mérünk.
A tökéletes gáz p. Vm szorzatát választották a hőmérsékletmérés alapjául. ennyiség adott között) körülmények Áll. nyomáson (izobár térfogata 1ºC hőm. emelkedés hatására a 0ºC-on mért térfogat(V 0) 1/273 -ad részének növekedésével jár
Áttérve az abszolút hőmérsékleti skálára: (T = 273, 15 +t) p Vm = R T p V=n R T R = 8, 314 Jmol – 1 K-1 A víz hármaspontjához rögzítik a termodinamikai hőmérséklet-skálát (a hármaspontban mindhárom halmazállapot egyszerre jelen van): 1 Kelvin (K) egyenlő a víz hármaspontja hőmérsékletének 1/273, 16 részével. A víz hármaspontja tehát pontosan 273, 16 K
Nyomás p = F/A (nyomóerő/felület) Egysége: Pascal (N/m 2) 1 bar = 105 Pa 1 atm = 1, 013 105 Pa 1 torr = 1 Hgmm: 1 mm magas higanyoszlop hidrosztatikai nyomása (p = g h) Gázokban a nyomás a molekulák által szállított impulzus áramsűrűsége. Erő:
fal mvx -mvx = 2 mvx (A zárójel időátlagolást jelent. ) Parciális nyomás: pi = xi p, pi = p (xi : móltört) Tökéletes gázokban pi az a nyomás, melyet a gáz akkor fejtene ki, ha egyedül töltené ki a rendelkezésre álló teret (Dalton törvénye).
Tökéletes és reális gázok Gázok: Az anyagi részecskék olyan halmaza, amelyre az jellemző, hogy: • nincs saját alakja, • kitölti a rendelkezésre álló teret, • gyenge kölcsönhatás a részecskék között
• térfogat V egységei: m 3, dm 3 (liter) mérése • anyagmennyiség (mól) n Avogadro-szám: 6, 022 × 1023 db (atom, molekula, ion, e-, foton. . . ) egysége: mol (mmol, μmol) az anyagmennyiség nem azonos a tömeggel (a tömeg a tehetetlenség mértéke és ennek SI mértékegysége a kilogramm)
Kísérleti tapasztalatok • kapcsolatok két állapotjelző között p 1/V (V 1/p) V = állandó × T p = állandó × T V = állandó × n Vm = V/n Boyle–Mariotte Gay-Lussac I. (Charles) Gay-Lussac II. Vm Avogadro móltérfogat • összevonásuk: p V = n R T a tökéletes gáz állapotegyenlete, gáztörvény
p. V=n. RT p 1/V vagy (V 1/p) (Boyle–Mariotte) p. V = állandó [ha n, T = áll. ] a görbe: izoterma alakja: hiperbola Határtörvény: kis p!
p. V=n. RT p = n R T (1/V)
V = (n R /p) T Gay-Lussac I. izobar p = (n R /V) T Gay-Lussac II. izochor
V = állandó × n Vm = V/n Vm = moláris térfogat (móltérfogat) p V = n R T vagy p Vm = R T ez az - egyesített gáztörvény vagy - a tökéletes gáz állapotegyenlete A tökéletes gáz egy „hipotetikus” anyagi minőség. Kis p-n, nagy T-n szinte minden gáz “tökéletes”.
Az állapotegyenlet ábrázolása háromdimenziós koordinátarendszerben: állapotfelület f (p, V, T, n) = 0
Gázelegyek: (többkomponensű rendszerek) A gázok korlátlanul elegyednek egymással. A tökéletes gázok ideális elegyet alkotnak. Dalton: a tökéletes gázelegy nyomása az egyes komponensek parciális nyomásának összege: p = p. A + p. B + … parciális nyomás: az a nyomás, amit a gáz egyedül fejtene ki az adott körülmények között:
móltört: az adott anyag mennyisége a mintában lévő összes anyagmennyiséghez viszonyítva xj = nj /n, ahol n = n. A + n. B + …, 0 < xj < 1. p = p. A + p. B p = x. A p + x. B p
Valóság: • a gázok cseppfolyósíthatók, ami arra utal, hogy a részecskék között kölcsönhatás van. • a gázrészecskéknek van saját térfogatuk, azaz a gáz nem nyomható össze végtelen kis térfogatúra.
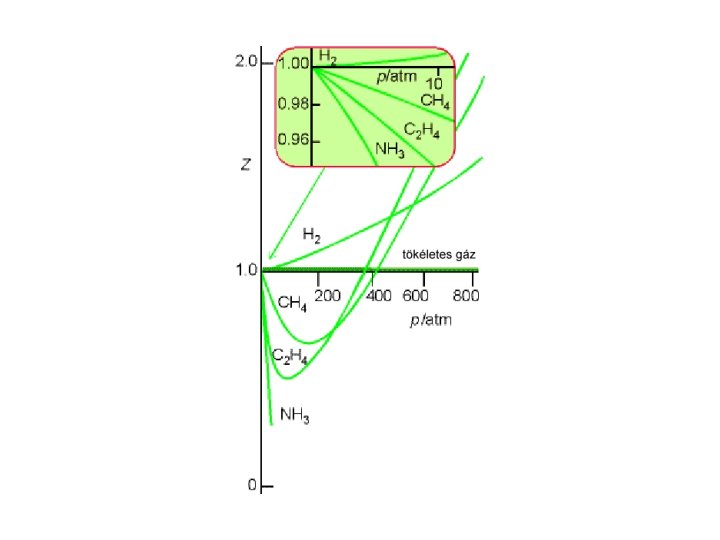
Reális gázok: az egyszerű egyenletek bizonyos körülmények (nagy p, kis T) között nem érvényesek. • kompresszibilitási együttható – Z: p. Vm/(RT) = Z - megmutatja az eltérést • viriálegyütthatós egyenletek: p Vm = RT (1 + B’p + C’p 2 + ··· ) • van der Waals-féle egyenlet(ek):
Az eltérés mértéke Tökéletes gázra: Reális gázra: p. Vm /RT = 1. p. Vm /RT = Z Z: kompresszibilitási együttható
Nem keresünk új egyenletet. Módosítjuk a p. Vm = RT egyszerű alakot. Az egyik matematikai módszer a viriál együtthatók alkalmazása: viriál állapotegyenlet p Vm = RT (1 + B’p + C’p 2 + ··· ) vagy p Vm = RT (1 + B/Vm + C/Vm 2 + ··· ) Ezzel a mért adatok pontosan visszaadhatók, de minden gázra, minden T- n B és C-t mérni kell! Csak az egyenlet alakja univerzális, a benne szereplő konstansok egyediek.
A másik módszer: a van der Waals-egyenlet A térfogat korrekciója: a molekulák sajáttérfogatával arányos b konstans. Vm helyett (Vm – b) lesz. A b értéke függ az anyagi minőségtől, de független a T-től. A nyomáskorrekció: a vonzó- és taszító hatásokból ered. p helyett (p + a /V m 2) lesz, ahol a értéke függ az anyagi minőségtől, de független a T-től.
A tökéletes gáz a reális gáz állapotfelülete
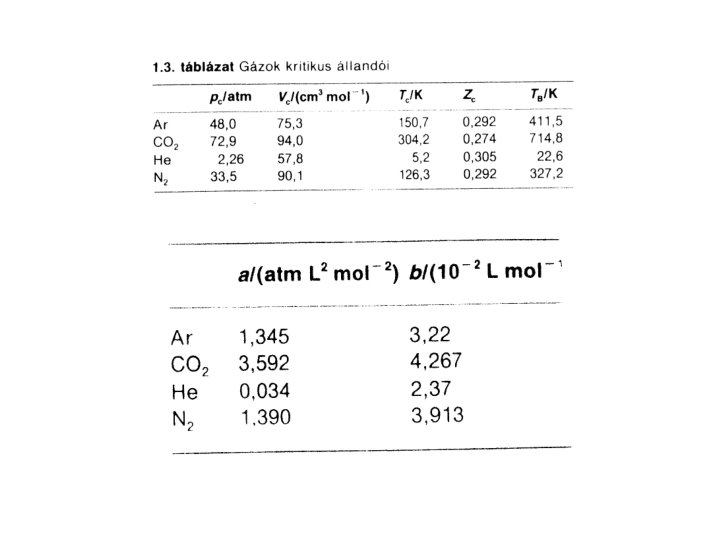
A reális gáz izotermái különböző hőmérsékleten: csökkentve T-t elérjük a kritikus állapotot – a gőz–folyadék határfelület elmosódik, eltűnik. A kritikus pont (*) jellemzői: a kritikus állapotjelzők Tc: kritikus hőmérséklet pc: kritikus nyomás Vm, c: kritikus móltérfogat
A kritikus állapotjelzők és a van der Waals konstansok (a, b) kapcsolata: a kritikus pont inflexiós pont, itt mindkét derivált (dp /d. Vm) = 0 és (d 2 p /d. Vm 2 ) = 0. Ebből: Vc = 3 b pc = a/27 b 2 Tc = 8 a/27 Rb Továbbá: kritikus kompresszibilitási együttható: Zc = pc Vc /R Tc = 3/8
A megfelelő állapotok tétele: bevezetjük a redukált állapotjelzőket pr = p/pc Vr = V/Vc Tr = T/Tc Ha a reális gázok redukált térfogata és hőmérséklete megegyezik, akkor azonos redukált nyomást fejtenek ki. Tr nő ↑
A termodinamika 0. főtétele 0. főtétel: izolált rendszer egyensúlyi állapot felé tart, a kialakuló egyensúly stabil egyensúly : nincsenek makroszkopikus áramok (<-> mikroszkopikus transzport lehet) nyílt zárt szigetelt izolált : környezetével bármilyen kölcsönhatás megengedett : felületén nincs anyagtranszport : felületén nincs hőátadás, csak munkavégzés : környezetével semmilyen kölcsönhatásban nincs az egymással egyensúlyban lévő rendszerek elláthatók címkékkel (állapotjelzőkkel) az intenzívek (pl. hőmérséklet, nyomás) jelzik két rendszer kölcsönös egyensúlyát.
A belső energia, a termodinamika I főtétele E = E kin + Epot + U Belső energia U: a rendszert felépítő részecskék kinetikus és potenciális energiájának az összege. Nem foglalja magában az egész rendszernek, mint makroszkopikus testnek a kinetikus és potenciális energiáját.
A belső energia részei: Termikus energia: az atomok, molekulák, ionok mozgásával kapcsolatos (haladó mozgás, rezgés, forgás) Intermolekuláris energia: a molekulák közötti erőkkel kapcsolatos (pl. folyadék elpárologtatásához energia kell, mert le kell győznünk a molekulák között ható vonzó erőket) Kémiai energia: a kémiai kötések létesítésével és felbontásával kapcsolatos. Magenergia: a nukleonok (protonok és neutronok) közötti kölcsönhatások energiája. A felosztást tovább folytathatjuk. Einstein: E = mc 2
Gyakorlatilag a belső energia abszolút értékét nem tudjuk pontosan megadni, csak a változását: U. Önkényes vonatkozási ponthoz viszonyítjuk (pl. 25 o. C, 1 bar nyomás - lásd „Entalpia” című fejezet) I. főtétel: Energia-megmaradás törvénye Elszigetelt rendszer: U = 0 Zárt rendszer: U = W + Q Infinitezimális változásra: d. U = W + Q Nyitott rendszer: lásd később. W: munka Q: hő
A munka A mechanikai munka az erő és az elmozdulás skalárszorzata: Termodinamikában a legtöbbet a térfogati munkával találkozunk.
Megjegyzések: a) Térfogati munkában mindig a külső nyomás (p. K) szerepel. reverzibilis változás: p = p. K b) Ha a térfogat nő, a munka negatív, ha csökken, pozitív. c) Ha a p állandó, könnyen integrálhatunk:
A térfogati munka szemléltetése: indikátordiagram aa b Állandó hőmérsékleten kiterjesztjük a gázt I. állandó térfogaton lehűtjük a gázt végső nyomásra II. állandó nyomáson felmelegítjük Wa Wb Tehát a térfogati munka útfüggvény.
A térfogati munkán kívül sokféle más munka is előfordul a termodinamikában. Az elemi munka egy intenzív mennyiség és egy extenzív mennyiség megváltozásának szorzata. Munka Intenzív m. Extenzív m. Elemi munka Térfogati Nyomás (-p) Térfogat V Határfelületi Fel. fesz. ( ) Felület (A) Elektrosztat. Potenciál ( ) Töltés (q) Wtér = - pd. V Wfe l= d. A Wel = dq A munka a rendszer határfelületén fellépő energiatranszport-mennyiség, amelyet a kölcsönhatáshoz tartozó (hőmérséklettől különböző ) intenzív állapotjelző inhomogenitása, a hajtóerő hoz létre.
A hő a rendszer határfelületén fellépő, anyagtranszport nélküli energiatranszport-mennyiség, amelyet a hőmérsékleteloszlás inhomogenitása hoz létre. Hőátmenettel járó folyamatok: A) Melegítés, hűtés B) Fázisátalakulás C) Kémiai reakció
A) Melegítés-hűtés: Q = c · m · T c = fajlagos hőkapacitás (fajhő) [J/kg·K] Víz c = 4. 18 k. J/kg·K Q = Cm · n · T Cm = moláris hőkapacitás (mólhő) [J/kg·K] A fenti egyenletek csak közelítő jellegűek. A hőkapacitás függ a hőmérséklettől:
A hő, mint a munka, útfüggvény. Ki kell jelölnünk az utat is. Leggyakoribb az állandó nyomáson vagy az állandó térfogaton végzett melegítés. Cmp>Cm. V , mert állandó nyomáson végzett melegítés során térfogati munkavégzés is van, a befektetett energiának azt is fedezni kell.
B) Fázisátalakulás A fázisátmenetek izoterm és izobár folyamatok. Tiszta anyag esetén vagy a hőmérsékletet vagy a nyomást választhatjuk szabadon. (Pl. a víz forráspontja 1, 013 bar nyomáson 1000 C). Olvadáshőt, párolgáshőt látens hőnek hívjuk (hőt közlünk, miközben nem nő a hőmérséklet). C) Kémiai reakció (ezzel később foglalkozunk)
Az entalpia Az első főtétel: Ha nincs munka ( V=0), a belsőenergia-változás a hővel egyenlő. (állandó térfogaton, ha egyéb munka sincs) Az állandó térfogaton lejátszódó folyamatokat jól jellemzi a belső energia. A kémiában nagyon gyakori az állandó nyomás. Ezért definiáltak egy olyan függvényt, amellyel az állandó nyomáson végbemenő folyamatokat jellemezhetjük.
Entalpia: Képezzük a teljes differenciálját. Ha csak térfogati munka lehet és a változás reverzibilis: Ekkor Ha a nyomás állandó: Mértékegysége: Joule Véges változásra: H = U + p. V állandó nyomáson: H = U +p v U = W + Q csak térfogati munka: W = -p V H = -p V + Q + p V H = Qp
Izobár folyamatban (ha nincs egyéb munka) az entalpiaváltozás a hővel egyenlő. Az entalpia-változás számítása izobár melegítés ill. hűtés esetén: Cmp hatványsorok formájában:
Fázisátalakulások: Hm (párolgás): - moláris párolgáshő Hm (olvadás): - moláris olvadáshő Az entalpia a környezetével mechanikai egyensúlyban lévő rendszer együttes energiáját adja meg. m A hh 0 p Az m tömegnek az edény aljához viszonyított helyzeti energiája:
A tökéletes gáz fogalma A tökéletes gáz jellemzői: 1. Nincs kölcsönhatás a molekulák között. 2. A molekula saját térfogata elhanyagolható az össz-térfogathoz viszonyítva. A tökéletes gázokra érvényes az általános gáztörvény: p. V = n. RT
Két molekula közötti potenciális energia a távolság függvényében Epot taszítás kis nyomás vonzás 0 r
Kisnyomású gázok megközelítik a tökéletes gáz vislekedését. Abból a feltételből kiindulva, hogy a molekulák között nincs pot. energia, következik, hogy a tökéletes gáz belső energiája nem változik meg, ha növeljük vagy csökkentjük a térfogatot (ill. a nyomást) állandó hőmérsékleten. Tökéletes gáz belső energiája csak a hőmérséklettől függ.
Entalpia: H = U + p. V csak a hőmérséklettől függ (Boyle – Mariotte törvény: állandó hőmérsékleten p. V = állandó) Tökéletes gáz entalpiája is csak a hőmérséklettől függ.
Összefüggés Cmp és Cmv között (tökéletes gáz) mert állandó nyomáson végzett melegítéskor kiterjed a gáz, és térfogati munkát végez
H = U + p. V = U + n. RT (tökéletes gáz)
Tökéletes gázok állapotváltozásai (izobár, izosztér, izoterm) A reverzibilis állapotváltozásokat tárgyaljuk. A gázok valóságos folyamatai nagyon jól megközelítik a reverzibilis folyamatokat. p 1 2 3 4 1 -2: izobár 3 - 4: izosztér 2 - 3: izoterm V
Izobár Térfogati munka: Hő (entalpia-változás): Belsőenergia-változás:
Izosztér (izochor) Térfogati munka: W=0 Hő (belsőenergia-változás): Entalpia-változás:
Izoterm U = 0 Q = -W Térfogati munka: Boyle-Mariotte törvény: H = 0
Hő Tök. gázok esetén tetszőleges folyamatban: U állapotfüggvény. A folyamatot gondolatban két lépésben hajtjuk végre I. izoterm (kiterjesztés V 2 -re) II. izosztér (melegítés T 2 -re) U = UI + UII UI = 0
Hasonlóan bizonyítható, hogy tökéletes gázban tetszőleges folyamatra: Tökéletes gázok reverzibilis állapotváltozásai:
Tökéletes gázok adiabatikus reverzibilis állapotváltozása Adiabatikus: Q = 0, U = W Kompresszió (összenyomás): a gázon végzett munka a belső energiát növeli - felmelegedés Expanzió (kiterjedés): a gáz munkát végez a belső energia rovására - lehülés Adiabatikus folyamatban mindhárom állapotjelző (p, T, V) változik.
A p - V diagramon az adiabata meredekebb, mint az izoterma. p adiabata T 2 T 1 V
Az adiabata egyenletének levezetése a) V és T kapcsolata Itt vezetjük be azt a feltételt, hogy a folyamat reverzibilis (tökéletes gáz) Integráljuk a kezdeti (1) és a végállapot (2) között. Cmv hőmérséklet-függésétől eltekintünk.
Cmv-vel átosztunk (Poisson-állandó)
P és V, ill. p és T kapcsolatának megállapításához a tökéletes gáz állapotegyenletét használjuk fel (p. V = n. RT). b) p és V kapcsolata
c) p és T kapcsolata
A standard reakcióhő Melegítéskor és hűtéskor a belső energiának elsősorban a termikus, másodsorban az intermolekuláris energia része változik. Kémiai reakciókban a kémiai kötésekben rejlő energiák változnak meg. Példa: a 2 H 2 + O 2 = 2 H 2 O reakcióban a H-H és az O-O kötések felszakadnak, és O-H kötések jönnek létre. Exoterm (hőtermelő) reakcióban energia szabadul fel. Endoterm (hőtemésztő) reakcióhoz energiára van szükség.
Exoterm (hőtermelő) és endoterm (hőemésztő) reakciók
Reakcióhő A reaktor hőmérséklete megegyezik a környezetével. A reakció során vagy hőt vesz fel a rendszer (endoterm reakció), vagy hőt ad le (exoterm reakció). Q T Endoterm: Q > 0 T Reaktor Exoterm: Q < 0
Reakcóhőnek nevezzük az állandó hőmérsékleten a reakcióegyenlet által definiált mennyiségű átalakulás során elnyelt vagy felszabadult hőmennyiséget. Állandó térfogaton: r. U, állandó nyomáson: r. H Példa: 2 H 2 + O 2 = 2 H 2 O 2 mól (4 g) hidrogén reagál egy mól (32 g) oxigénnel és 2 mól (36 g) víz keletkezik r. U = 2 Um(H 2 O) - 2 Um(H 2) - Um(O 2) r. H = 2 Hm(H 2 O) - 2 Hm(H 2) - Hm(O 2) Az így definiált reakcióhő függ a hőmérséklettől, a nyomástól, valamint a kiindulási anyagok és a termékek koncentrációjától.
Standardizálás során rögzítjük a nyomást és a koncentrációt. A standard reakcióhő reakcióegyenlet által definiált mennyiségű átalakulás során elnyelt vagy felszabadult hőmennyiség, miközben po = 105 Pa nyomású tiszta reagensekből ugyanilyen nyomású és azonos hőmérsékletű tiszta termékek kelekeznek. A standardizálás tehát : 1. tiszta komponenseket 2. po nyomást jelent A hőmérséklet nincs rögzítve (bármely hőmérsékleten beszélhetünk standard reakcióhőről), de a legtöbb adat 298 K-en áll rendelkezésre.
A továbbiakban a standard állapot jelölése: a felső indexbe írt 0 Standard nyomás: 0 5 p (=10 Pa = 1 bar)
Az entalpia jelentéséből ( H = Qp ) következik, hogy a standard reakcióhő lényegében entalpia-változás. Általános reakció: n. AMA = n. BMB n: sztöchiometriai együttható, M: molekulák, A a kiindulási anyagok, B a termékek indexe. A standard reakcióhő (standard reakcióentalpia): a standard moláris entalpia.
Példa: 2 H 2 + O 2 = 2 H 2 O Meg kell adni a reakcióegyenletet, valamint a résztvevő anyagok halmazállapotát. Példák: 2 H 2(g) + O 2 (g)= 2 H 2 O(l) H 2(g) + 1/2 O 2 (g)= H 2 O(g) Standard reakcióhő 25 o. C-on -571, 6 k. J -285, 8 k. J -241, 9 k. J
A reakcióhő mérése A reakcióhő mérésére használt eszköz a kaloriméter. Bombakaloriméter: elsősorban égéshő mérésére alkalmas. Az anyagot nyomásálló edényben (bomba) oxigénfeleslegben elégetjük.
Bombakaloriméter hőmérő Gyújtószerkezet Fűtőspirál keverő hőszigetelés
A reakcióhő meghatározható a hőmérséklet-emelkedésből ( T): q = C· T, C a kaloriméter hőkapacitása (minden, ami a hőszigetelésen belül van, edény fala, víz, bomba, stb). C meghatározása: ismert mennyiségű elektromos energiával, amely T´ hőmérséklet-emelkedést okoz: U·I· t = C· T´, ahol U a feszültség, I az áramerősség, t a melegítés időtartama.
A bombakaloriméterben Dr. U-t mérünk, mert a térfogat állandó. H = U +p. V r. H = r. U + r(p. V) A p. V szorzat elsősorban a gáz halmazállapotú anyagok molekulaszám-változása miatt változik. Tökéletes gáz közelítés: p. V = n. RT. Eszerint r(p. V) = rng. RT, ahol rng a reakció során a gáz halmazállapotú komponensek sztöchiometriai koefficienseiben bekövetkező változás: rng = ng(termékek) - ng(reaktánsok)
Példa: C 6 H 5 COOH(s) +7, 5 O 2 = 7 CO 2(g) +3 H 2 O(l) rng= 7 - 7, 5 = -0. 5 Az eltérés r. U és r. H között általában nem jelentős.
Hess tétele Az entalpia állapotfüggvény: változása kizárólag a kezdeti és végállapottól függ (független a közbülső állapotoktól). A megállapítás alkalmazható a reakcióhőre: A reakcióhő független attól, hogy a reakció milyen közbülső termékeken keresztül megy végbe.
Példa: A C(grafit) + O 2 = CO 2 (1) reakció entalpia -változása megegyezik az alábbi két reakció entalpiaváltozásának az összegével: C(grafit) + 1/2 O 2 = CO (2) CO +1/2 O 2 = CO 2 (3) r. H(1) = r. H(2) + r. H(3) Így ha a három reakcióhő közül kettőt ismerünk, a harmadik kiszámítható.
Hess 1840 -ben kísérleti tapasztalatok alapján állította fel tételét. A Hess-tétel jelentősége: Nehezen vagy egyáltalán nem mérhető reakcióhőket is meghatározhatunk számítással.
Reakcióhő számítása égéshőkből: Kiszámíthatjuk a reakcióhőt, ha ismerjük minden résztvevő égéshőjét. Gondolatban a kiindulási anyagokat elégetjük, majd az égéstermékekből fordított égési folyamattal előállítjuk a termékeket. c. H: égéshő (c a „combustion” = égés rövidítése) Égéstermékek n. A c. HA Kiind. anyagok - n. B c. HB Termékek
A reakcióhőt tehát megkapjuk, ha a kiindulási anyagok égéshőinek összegéből kivonjuk a termékek égéshőinek összegét: r. H = - r( c. H) Példa: 3 C 2 H 2 = C 6 H 6 r. H = 3 c. H(C 2 H 2) - c(C 6 H 6)
A képződéshő az elemekből (pontosabban az elemeknek az adott hőmérsékleten legstabilabb módosulataiból) végbemenő képződési reakcióhője. Jele f. H (f a „formation” = képződés rövidítése). Példa: Az SO 3 standard képződéshője az S +3/2 O 2 = SO 3 reakció standard reakcióhője. A definícióból következik, hogy az elemek képződéshője 0 (bármely hőmérsékleten).
Reakcióhő számítása képződéshőkből: Képzeletben a kiindulási anyagokat először elemeire bontjuk (a képződés fordítottja), majd az elemekből összerakjuk a termékeket. Elemek - n. A f. HA Kiind. anyagok n. B f. HB Termékek
A reakcióhőt tehát megkapjuk, ha a termékek képződéshőinek összegéből kivonjuk a kiindulási anyagok képződéshőinek összegét: r. H = r( f. H) Példa: 3 C 2 H 2 = C 6 H 6 r. H = f. H(C 6 H 6) - 3 f. H(C 2 H 2)
Standard entalpiák A belső energiának és az entalpiának nem kíséreljük meg az abszolút értékét meghatározni. Nemzetközi megállapodás rögzíti az elemek és vegyületek standard entalpiáját.
1. 298, 15 K-en (25 o. C-on) és po = 105 Pa nyomáson az elemek stabilis módosulatának az entalpiáját 0 -nak vesszük: (elemek)
25 o. C-tól eltérő hőmérsékleten már nem 0 az entalpia. Pl. 25 o. C-on szilárd, T hőmérsékleten gáz halmazállapotú elem standard moláris entalpiája T Kelvinen: olvadáspont Szilárd anyag moláris hőkapacitása forráspont Szilárd anyag Folyadék olvadáshője moláris hőkapacitása Folyadék párolgáshője Gőz moláris hőkapacitása
2. A vegyületek standard entalpiáját 298, 15 K-en azonosnak vesszük a standard képződéshőjükkel. De csak 298 K-en! Minden más hőmérsékleten eltér az entalpia a képződéshőtől. Táblázatokban: standard entalpiák 298 K-en és moláris hőkapacitás (Cmp) függvények
Standard reakcióhő kiszámítása T K-en: 1. Kiszámítjuk az összes résztvevő standard entalpiáját T K-en. 2. Képezzük a különbséget.
Nyitott rendszer energiamérlege, stacionárius rendszerek Környezettel anyag-és energiacsere is megengedett. A technológiai folyamatok általában nyitott rendszerek.
Az első főtétel zárt rendszerre: U = Q + W Nyílt rendszer A belépő és kilépő anyagok energiát visznek magukkal (Ube - Uki). A mozgatásuk is energia-felhasználással jár. (a bejuttatás energiáját pozitív, a távozásét negatív előjellel vesszük figyelembe).
Q lbe pbe W rendszer lki pki Abe Aki Az anyagok be- és kijuttatását egy-egy dugattyús hengerrel szimbolizáljuk. Vbe Vki U = Q + W + Ube - Uki + pbe. Abelbe - pki. Akilki U = Q + W + Hbe - Hki Ez az I főtétel nyitott rendszerre
A stacionárius (állandósult) rendszer olyan nyitott rendszer, amelyben az állapotfüggvények függnek a helytől, de időben nem változnak. Energia nem fogy és nem halmozódik fel: DU = 0 Hki - Hbe = Q + W (Stacionárius reaktor entalpiamérlege) Össz kivitt entalpia Össz Hő bevitt entalpia Munka
Ha nincs reakció, Hki - Hbe az áthaladó anyag entalpiaváltozása: DH = Q + W Három fontos példa, amely a műszaki gyakorlatban előfordul: 1. Fojtószeleppel gázok nyomását csökkentjük. p 2 p 1 p 2 > p 1 DH = 0 Folytonos működésű, a be- és kilépő gáz állapotjelzői időben állandóak. Adiabatikus a folyamat: Q = 0 Nincs munkavégzés: W= 0.
2. Folytonos adiabatikus kompresszor Q = 0, DH = Wk Wk : a kompresszor gépi munkája 3. Stacionárius reaktor ( Hki - Hbe =) nki. Hmki - nbe. Hmbe = Q + W