Identificacin de Elementos Reguladores Permanentes Mediante Anlisis Comparativos


































- Slides: 35
Identificación de Elementos Reguladores Permanentes Mediante Análisis Comparativos del Genoma Nombre: Fco. Javier Rodríguez Blázquez D. N. I. : 75152911 C Titulación: Ing. Informática.
Introducción La información en los genes fluye desde las secuencias estáticas de ADN hasta la activación de las proteínas mediante un ARN que actúa como intermediario. Los genes se activan o reprimen selectivamente gracias a la actuación de secuencias reguladoras dentro del ADN. ¿Selectivamente? La regulación depende de diversos factores fisiológicos, medioambientales y del desarrollo del contexto celular.
En el genoma humano las regiones que se encargan de regular la trascripción genética están compuestos por lugares de unión de donde los factores de trascripción llegan para unirse a la secuencia. Un factor de transcripción es una proteína que participa en la iniciación de la transcripción del ADN, pero que no forma parte de la ARN polimerasa. Los factores de transcripción pueden ser activados o desactivados selectivamente por otras proteínas, a menudo como paso final de la cadena de transmisión de señales intracelulares. Los complejos de transcripción en las células eucarióticas son mucho más complejos que en procariotas debido al mayor tamaño del genoma eucariótico, por lo que el complejo de transcripción en eucariotas necesita un mayor número de etapas para ensamblarse. Las ARN-polimerasas son un conjunto de proteínas con carácter enzimático capaces de polimerizar los ribonucleótidos para sintetizar ARN a partir de una secuencia de ADN que sirve como patrón o molde. La ARN polimerasa más importante es la implicada en la síntesis del ARN mensajero
Si conocemos la identidad del factor de transcripción Podemos conocer la función del gen La mayoria de los organismos eucariotas toleran considerables variaciones en la secuencia en las zonas de llegada Un simple consenso no sirve para hacer una aproximación representativa de los factores de trascripción Se utiliza una matriz de posiciones ponderadas mediante pesos para representar cuantitativamente la exactitud y especifidad de la unión !!!!
Las matrices pueden ser muy precisas en identificar secuencias de unión in vitro pero no identifican con exactitud los sitios con funciones in vivo que dan predicciones muy significativas La especifidad de unión de un factor de transcripción in vivo depende de propiedades adicionales que no se modelan con una matriz de pesos como: 1. Interacción proteína-proteína 2. Concentración de factores de trascripción In vitro: In vivo: Método de investigación que utiliza seres vivos, ej: pruebas en animales. Este tipo de investigación da una visión general del efecto causado por el experimento Método de investigación que se origina en un tubo de ensayo y generalmente fuera del organismo vivo. Se trabaja con células, órganos…Se utiliza para investigar mecanismos de acción y cuenta con pocas variables
La comparación de genes ortólogos es una herramienta poderosa para el análisis del genoma. Llamamos genes ortólogos a los que son semejantes por pertenecer a dos especies que tienen un antepasado común Phylogenetic Footprinting Es una técnica que identifica elementos regulatorios buscando regiones inusualmente conservadas en un congundo de secuencias ortólogas de ADN no codificante de múltiples especies
Con. Site Servicio disponible en la Internet que implementa algoritmos para la identificación de regiones regulatorias mediante el análisis comparativo de secuencias. Alinear y analizar los sitios conservados mediante la comparación de dos secuencias de ortólogos Analizar secuencias individualmente
Con. Site Este algoritmo para la detección de zonas de unión de factores de transcripción (TFBSs) está basado en las técnicas Phylogenety Footprinting. Tiene tres componentes principales: Conjunto no redundante de modelos de unión de los factores de transcripción 1 2 Un algoritmo de alineamiento apropiado para secuencias no codificantes de ortólogos Software dividido en módulos para aunar las predicción de zonas de unión con los análisis de similaridad de las secuencias 3
Resultados Se pueden identificar potenciales zonas de unión de los factores de trascripción dentro de secuencias genómicas mediante aproximaciones muy bien estudiadas basadas en los perfiles cuantitativos que describen sitios de unión característicos para los factores de traducción Numero de zonas de unión bioquímicamente determinadas La calidad de los modelos de matrices ¿Cómo? In vivo Pocos pero significativos sitios de unión In vitro Muchos pero menos expresivos
Mezclamos los datos recolectados de ambos métodos para crear un conjunto de 108 perfiles de zonas de unión no redundantes de alta calidad Los perfiles se derivan normalmente de vertebrados, insectos y plantas pero la gran mayoría (65%) se corresponde con modelos de matrices de unión de factores de humanos y de roedores
Prediciendo sitios de unión Analizando la conservación en secuencias ortólogas Phylogenetic Footprinting proporciona información complementaria a las obtenidas en la predicción de los sitios de unión para los análisis de la regulación de los genes Hipótesis Las secuencias que codifiquen funciones importantes estarán sometidas a una elevada presión para que se mantengan constantes y no se modifiquen a lo largo de periodos de evolución razonables
La clasificación de las secuencias como secuencias preservadas en el tiempo o libremente evolucionadas aún no es un proceso cuantitativo. Secuencia original ? Secuencia conservada Secuencia evolucionada Debe señalarse que el grado de evolución varía dramáticamente genes y que la elección de la especies es un elemento muy importante a tener en cuenta en los estudios de Phylogenetic Footprinting. Una distancia evolutiva demasiado grande puede originar alteraciones en la forma de regulación de los genes que se estan estudiando
Una distancia evolutiva insignificante no mejora significativamente la exactitud total de las predicciones. Hemos desarrollado el método Con. Site para integrar la técnica de Phylogenetic Footprinting con las predicciones basadas en los modelos de zonas de unión para los factores de trascripción, a fin de llevar a cabo predicciones específicas y concluyentes de elementos reguladores de funciones. 17 -20 % En este informe, nos centramos en la comparación humano-roedor, varios estudios han sugerido que sólo un pequeña parte (17 -20%) de regiones no codificantes se conservan (de media) a esta distancia de evolución
Las secuencias están salpicadas con segmentos distinguibles de alta similaridad flanqueado con regiones de secuencias aparentemente aleatorias (aproximadamente el 33% de identidad nucleótida está observada entre secuencias genómicas aleatorias, con extensas variaciones dependiendo del algoritmo de alineamiento utilizado, las configuraciones iniciales y las características de la secuencia). Este modelo de `similitud separada en compartimentos’ es consistente a el descubrimiento de múltiples factores de transcripción unidos a grupos localmente muy densos llamados módulos regulatorios , que sugieren que los distintos bloques de la secuencia son requeridos para la regulación transcripcional Secuencia 1 Secuencia 2
A fin de identificar segmentos que se conservan en secuencias de ortólogos, deben ser definidos un apropiado conjunto de criterios de clasificación. Como la similaridad o la tasa de evolución varían extensamente a través de las secuencias genómicas, un único umbral no va a funcionar correctamente. El algoritmo se centra en segmentos de similaridad elevada. Esto se refiere a que las ventanas correderas tienen un tamaño fijado sobre el alineamiento, reteniendo solo en aquellas donde la identidad en la secuencia excede de un umbral por defecto o definido por el usuario. Si una secuencia de ADN complementario esta disponible, el programa de análisis puede excluir de las consideraciones a las predicciones de sitios unidos situados dentro de exones presentes en el alineamiento de secuencias genómicas.
El ADN complementario o ADNc es un ADN de cadena sencilla. Se sintetiza a partir de una hebra simple de ARNm maduro. Se suele utilizar para la clonación de genes eucariotas en células procariotas. El dogma central de la biología molecular dice que durante la síntesis de proteínas, el ADN es transcrito en ARNm, que a su vez es traducido a proteínas. Una diferencia entre ARNm eucariótico y procariótico es que el ARNm eucariótico puede contener intrones, secuencias no codificantes que deben ser extraídas del ARNm antes de ser traducido a proteínas. El ARNm procariótico no tiene intrones, así que no sufre ningún proceso de corte (splicing).
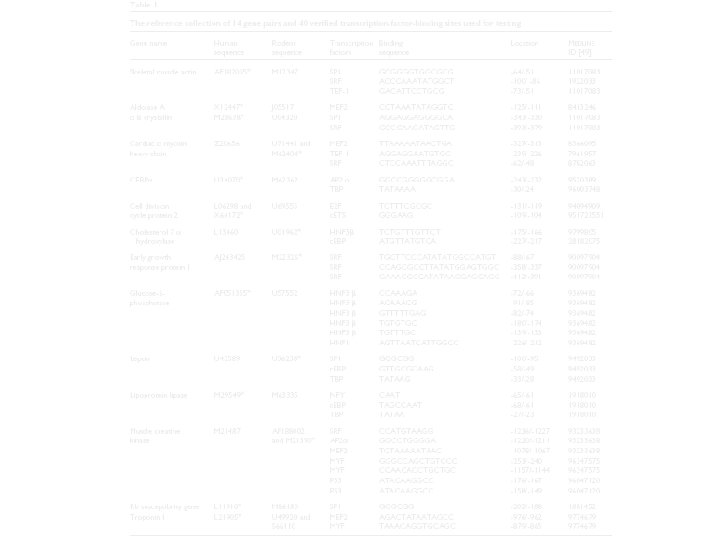
Evaluando el impacto de PF en la especificación de predicciones de sitios de unión. A fin de evaluar cuantitativamente la contribución de los análisis de comparación de secuencias a la exactitud de las predicciones de sitios de unión de los factores de trascripción, se adjunta un conjunto de 14 genes bien estudiados. Hemos comparados la selectividad de las predicciones de TFBS entre TFBS generados con secuencias aisladas de humano y TFBS generados con las mismas secuencias de genes filtrados mediante un análisis comparativo con la secuencia de genes ortóloga de ratón. Los pares de secuencias están limitados en longitud entre 680 y 2900 pares de bases, pero todas incluyen la region -500 a la +100 relativa al sitio de comienzo de la trascripción. Dentro de las 14 pares de secuencias hay 40 TFBSs experimentalmente definidos para 13 factores de trascripción distintos dentro de un conjunto de matrices disponibles. Para mayor claridad, estos sitios de unión no fueron utilizados en la construcción de los modelos matriciales. El corte de conservación fue colocado al 70% de todos los test, mientras que el tamaño de ventana para el análisis de conservación fue colocado en 50 pb.
Selectividad La cantidad de verdaderas zonas de unión de factores de transcripción dentro de la secuencia genómica va a ser pequeño (dicho por anticipado). Definimos la tasa falsos-positivos como el total de numero de predicciones de todos los modelos divididos por la longitud de la cadena de entrada. El número de zonas de unión de factores de trascripción predecidas fueron determinadas experimentalmente aumentando progresivamente los umbrales de las matrices de puntuación relativa entre el 65% y el 90% para ambas secuencias por separado y la correspondiente pareja de ortólogos. M = Conjunto de 108 modelos Pm, c= numero de zonas predichas utilizando el modelo m y el umbral de la matriz de puntuación relativa c L= longitud de la secuencia analizada (en pb)
La selectividad predecida (medida por el número medio de TFBSs predecidos por cada 100 pb de la secuencia promotora cuando se inspeccionaron todos los modelos) mejoro en un 85% cuando la técnica de Phylogenetic Footprintig fue aplicada. Las tasas de las puntuaciones de la selectividad observada utilizando Phylogenetic Footprintig y las tasas de las puntuaciones obtenidas utilizando modelos de análisis separado de secuencias se muestra a continuación
Los candidatos a zonas de unión para los factores de trascripción en una secuencia individual tienen una puntuación determinada por su posición en la matriz de pesos dada por la secuencia. Los rango de puntuación son únicos para cada modelo de unión, podemos convertir este rango a una escala relativa mediante la siguiente ecuación: Los rangos de puntuación se utilizan para crear los umbrales de las matrices de puntuación relativa
Sensibilidad La sensibilidad mide la habilidad para detectar correctamente sitios conocidos (esto es, cuando una predicción y un TFBSs ya anotado se superpone en por lo menos un 50% de la anchura, dando la correspondiente característica de unión para el factor de trascripción. Aumentando progresivamente los umbrales de las matrices de puntuación relativa entre el 65% y el 90% para ambas secuencias por separado y la correspondiente pareja de ortólogos vemos que a sensibilidad total (la cantidad de sitios conocidos que se han detectad) se redujo escasamente : un 65, 5% fue detectada con Phylogenetic Footprinting en comparación con el 72, 5% cuando se analizó las secuencias por separado.
El hecho de que unos pocos sitios no fueran detectados puede atribuirse a múltiples causas. Por ejemplo: 1. Zonas de unión de factores de trascripción pudieron no haberse conservado 2. Zonas de unión de factores de trascripción pudieron estar presentes no fueron detectados bajo los umbrales. 3. Concluimos que la mayoría de los sitios de unión anotados 4. experimentalmente se localizaron dentro de las regiones 5. conservadas, como pudimos detectar correctamente un 82, 5% 6. de los TFBSs con una puntuación de umbral del 60%, utilizando pares de genes ortólogos.
En Conclusión…
La comparación de secuencias de ortólogos es un método efectivo para identificar segmentos que probablemente intervengan en regulación una función biológica concreta. Los métodos basados en Phylogenetec Footprinting para identificar las zonas donde se unen los factores de trascripción dependen de: 1. El algoritmo de alineamiento 2. Los perfiles de unión disponibles 3. La distancia evolutiva de las secuencias comparadas
Hemos introducido dos nuevos conceptos: 1. La capacidad de recopilar un nuevo conjunto de características de las zonas de unión de los factores de trascripción a partir de investigaciones previas. 2. Un conjunto de elementos que determinan la bondad de los métodos basados en la técnica Phylogenetec Footprinting. Con. Site es una interfaz web que ofrece muchas facilidades de uso, característica indispensable para haces más ameno el trabajo de los investigadores que estudien a fondo diversos genomas.
La colección los perfiles de las zonas de unión es un recurso muy importnte para los proyecto bioinformáticos. La base de datos de perfiles JASPAR esta disponible de forma gratuita para todos los miembros de la comunidad científica. Los perfiles no son redundantes, pero están limitados a aquellos casos en los que hay suficiente información acerca de la unión como para generar una representación significativa de la unión exacta del factor de trascripción. La base de datos está en continuo crecimiento, debido a la investigación constante para determinar con exactitud las características de lossitios de unión del ADN ¿ ? Qué es JASPAR es un base de datos que contiene un conjunto de modelos de unión derivados en la mayor parte de aquellos conjuntos de modelos zonas de unión de factores de trascripción definidos para células eucariotas.
El nuevo conjunto de característica de las zonas de unión de los factores de trascripción que está basado en Phylogenetic Footprinting permite la valoración cuantitativa para desarrollar nuevos métodos. Estamos antes la colección más grande disponible para uso general de esta índole.
En nuestro el estudio detectamos que el 68% de las zonas de unión de los factores de trascripción experimentalmente definidos se encuentran en aquellos segmentos que han permanecido constantes (con un umbral de la matriz de puntuaciones relativas del 65%) En otro estudio acerca de las características de conservación relacionadas con las zonas de unión de los factores de trascripción bajaron el porcentaje a un 50% ¿ Por qué ?
1. Los procedimientos para generar las secuencias son diferentes. Por ejemplo, la cantidad de secuencia flanqueante usada para mapear la localización de los sitios en las secuencias fueron más bajas que en los estudios realizados previamente. 2. Los algoritmos de alineamiento son diferentes, con el antiguo conjunto alinebamos con BLAST y evaluavamos con un umbral de similaridad muy severo (>80% de identidades por cada 40 pb). 3. No se se excluyen pseudogenes o genes parálogos en el estudio previo, lo que puede decrementar la sensibilidad debido la aplicación errónea de la Phylogenetic Footprinting en genes que evolucionaron bajo condiciones de presión diferentes. genes parálogos, que son aquellos que se encuentran en el mismo organismo, y cuya semejanza revela que uno procede de la duplicación del otro.
Un pseudogén es una secuencia nucleótida similar a un gen normal pero que no da como resultado un producto funcional. Se han propuesto varios escenarios para explicar el origen de un pseudogen: Fragmentos de la transcripción en ARN mensajero de un gen pueden ser transcritos inversamente de manera espontánea e insertados en el ADN cromosómico (llamado retrotransposición). A estos pseudogenes se les llama procesados. Ya que estos pseudogenes carecen de los promotores de los genes normales, no se expresan con normalidad. Un suceso de duplicación genética puede significar que un genoma tenga dos copias de un gen cuando sólo necesita de una. Las mutaciones que desactiven una de las copias no serían, por tanto, seleccionadas en contra (e incluso podrían tener cierta ventaja selectiva estando desactivados). Además, el suceso de duplicación puede no ser completo, de manera que la copia tenga promotores incompletos. Estos pseudogenes se llaman duplicados o sin procesar. Un gen puede dejar de ser funcional o desactivarse si una mutación así se fija en la población. Esto puede ocurrir por medios normales como la selección natural o la deriva genética. Es el mismo mecanismo por el que los genes sin procesar se desactivan.
El trabajo que hemos presentado se concentra en las comparaciones con secuencias de mamíferos, pero no hay limitaicones dentro del sistema Con. Site. No excluye la posibilidad de estudiar otro tipo de organismos( insectos y nematodos ). Está caracteristica será importante e un futuro para desarrollar métodos capaces de analizar múltiples secuencias en paralelo, que no es nada trivial. Para preservar la sensibilidad es necesario elaborar métodos para ponderar las distancias evolutivas, que requiere avances en los algoritmo de alineamiento de múltiples secuencias.
Ningún recurso ofrece el amplio conjunto de funciones que Consite nos proporciona hoy en día. El único que puede hacerle sombra es el recientemente publicado r. Vista, que busca zonas de unión de los factores de trascripción en una secuencia de referencia y filtra los resultados para las zonas en regiones con de alta posibilidad de permanecer inalterables respecto a una segunda secuencia genómica. A diferencia de r. Vista, Con. Site busca en ambas secuencias TFBs para una mayor exactitud, y permite fáciles modificaciones de los parámetros en los análisis interactivos, además proporciona diferentes formatos de salida para ayudar al diseño y la interpretación de experimentos en biologia molecular. Secuencia de referencia Secuencia de consulta 1 Secuencia de consulta 2
Consite publica un conjunto de perfiles de factores de trascripción que permiten a los usuarios acceder a la información de los factores de trascripción asociados con las zonas que han predicho es sus estudios. También permite que los usuarios definan sus propios perfiles. Hemos presentado un algoritmo que utiliza Phylogenetic Footprinting para buscar potenciales TFBS. Los métodos anteriores, que se basaban únicamente en la búsqueda de un perfil en una sola secuencia genética proporcionaban una exactitud muy inferior a la que obtenemos identificando elementos regulatorios gracias a los métodos basados en la técnica Phylogenetic Footprinting.
En Resumen… Utilizando Phylogenetic Footprinting para filtrar las predicciones hechas computacionalmente se reduce el ruido significativamente en contra de disminuir escasamente la sensibilidad. La aplicación web Con. Site permite a los investigadores utilizar estos métodos sin coste alguno, totalmente gratis (y por lo tanto, legal). En un futuro… Cuando se termine de secuenciar el genoma completo del humano y del ratón este nuevo algoritmo será muy significativo para describir las funciones que se realizan en secuencias no codificantes.