Genetica Umana 20172018 Fulvio Cruciani Organizzazione del genoma



 - regione sesso specifica")





 • ANALISI Ipotesi: una mutazione Y-linked è avvenuta")
 L’analisi di questa regione attraverso una serie di tecniche")
 Am J Hum Genet 92: 301")


 §Cloni")
 ü Bassa densità genica (1100 geni, 7 geni/Mb):")
 Secondo l’ipotesi di Ohno (1967) il")
 T La maggior parte dei geni")


 all’estremità dei bracci")
 3. 1 1, 2% (coding)")


 Eteroplasmia: presenza di molecole di mt. DNA differenti in un")
 Il livello di eteroplasmia è inoltre differente tra cellule della")
 Variazione del numero mitocondri e del mt. DNA nel lifetime")



 Mutazioni nel gene per mt-t.")
 La più comune patologia mitocondriale primaria e presenta")

- Slides: 34
Genetica Umana 2017/2018 – Fulvio Cruciani Organizzazione del genoma umano “Cromosomi speciali” (X, Y, mt. DNA) Materiale didattico: - Strachan and Read (2012) capitolo 9 e 10. 3 - Schon et al. (2012) Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet 13: 878 -888 (esclusi box e mt. DNA nei tumori) Approfondimenti (facoltativi): - Skaletsky et al. (2003) The male-specific region of the human Y chomosome. Nature 423: 825 -837 - Wang et al. (2013) Genetic basis of Y-linked hearing impairment. Am J Hum Genet 92: 301 -306 - Ross et al. (2005) The DNA sequence of the human X chromosomes. Nature 434: 325 -336
Cromosomi “speciali”
Cenni sull’ evoluzione dei cromosomi sessuali
Cromosoma Y umano (2003) - regione sesso specifica
Le palindromi del cromosoma Y Elevata omologia di sequenza tra i due bracci delle singole palindromi: Duplicazione recente? No! Elevata omologia dovuta a conversione genica intracromosomica
Le palindromi del cromosoma Y: Conversione genica intracromosomica Elevata omologia di sequenza tra i due bracci delle singole palindromi dovuta a conversione genica non allelica intracromosomica. La conversione genica è un’importante meccanismo coinvolto nella riparazione dei DSBs che può portare a mutazioni Conversione genica: trasferimento unidirezionale di informazione genetica da una sequenza donatrice ad una sequenza accettore. Può essere allelica o non-allelica; intra o intercromosomica. Nel caso specifico (cromosoma Y) è non-allelica, intracromosomica e tra bracci di una palindrome
Delezioni tra ripetizioni dirette del cromosoma Y sono alla base della maggior parte dei casi di azoospermia Sono state definite tre regioni principali del cromosoma Y (AZFa, AZFb ed AZFc) la cui delezione porta a riduzione o mancata produzione di spermatozoi. Le regioni di omologia in AZFb ed AZFc (in figura) sono interne a palindromi differenti (P 5 e P 1; P 3 e P 1, rispettivamente). AZFa è invece delimitata da due elementi HERV AZFa = 0. 8 Mb AZFb = 5. 5 Mb AZFc = 3. 5 Mb
Crossing over ineguale intercromatidico tra bracci di una palindrome porta a cromosomi Y dicentrici (ed acentrici) Alcuni casi di azoospermia sono dovuti a ricombinazione omologa ineguale intercromatidica tra bracci diversi di una determinata palindrome (riquadro D in figura). Si genera un acentrico ed un dicentrico, entrambi con duplicazioni e delezioni.
Ereditarietà Y-linked: Geni situati nella regione MSY del cromosoma Y, che determinino caratteri la cui penetranza è completa dovrebbero: -Essere presenti solo nei maschi, -Essere presenti in tutti i figli maschi di un individuo affetto -Le figlie femmine di individui affetti non solo saranno normali, ma non avranno figli affetti. • Anni 50: descritti 17 tratti fenotipici ad ereditarietà “putativa” Y-linked • Esempio famiglia Lambert “gli uomini istrice” (1700) • 1957 – Stern: non c’è nessun tratto chiaramente Y-linked • 2004 – Page: “pedigree analysis has yet to reveal a single Y-linked gene” • 2004: viene identificata una famiglia cinese con apparente sordità Y- linked
Ereditarietà Y-linked Wang et al. (2013) • ANALISI Ipotesi: una mutazione Y-linked è avvenuta dopo la separazione dei due “rami” principali del pedigree (affetti e non affetti) 1) Si decide di sequenziare la regione non ripetitiva del cromosoma Y di due individui imparentati appartenenti ai due rami (frecce in figura). Per confermare che siano effettivamente parenti, si analizzano prima 67 microsatelliti Ylinked. Il sequenziamento evidenzia 4 sostituzioni di basi, tre nel ramo “non affetto” ed una nel ramo affetto. Quest’ultima si trova al di fuori di qualsiasi gene annotato o regione funzionale e viene considerata come non causale. 2) L’analisi comparata della “profondità” del deep sequencing dei due soggetti porta all’identificazione nel cromosoma Y dell’individuo affetto di 3 tre regioni duplicate che comprendono parte della famiglia genica TSPY. Sordità Y-linked
Ereditarietà Y-linked • ANALISI 3) L’analisi di questa regione attraverso una serie di tecniche citogenetiche (fiber FISH, CGH, Tail. PCR) porta all’identificazione “inattesa” di una inserzione nel cromosoma Y di circa 160 kb del cromosoma 1. Questa regione comprende 6 geni (uno parziale) che normalmente si trova in una porzione del cromosoma 1 (DFNA 49, MIM%608372) precedentemente associata con sordità autosomica. Sordità Y-linked Wang et al. (2013) INTERPRETAZIONE: Lo stesso gene (o geni) sono alla base della sordità DFNA 49 e della sordità Y linked. L’analisi della sequenza della regione duplicata del cromosoma 1 sull’Y non ha evidenziato mutazioni potenzialmente dannose. La spiegazione più parsimoniosa per la sordità Y linked viene considerata la sensibilità all’aumentato dosaggio di uno o più dei geni della porzione di crom. 1 duplicata.
Ereditarietà Y-linked Sordità Y-linked Wang et al. (2013) Am J Hum Genet 92: 301 -306
Coinvolgimento del cromosoma Y nei tumori e nell’invecchiamento? Science 2 January 2015 Am J Hum Genet 2017 LOY Loss Of Y chromosome
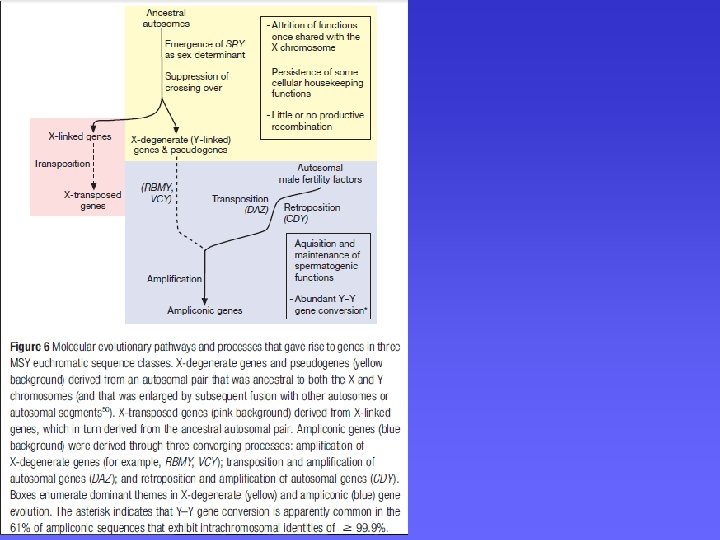
Cromosoma Y: difficoltà nel sequenziamento • Le strategie di mappatura applicate ad altri cromosomi non si sono rivelate efficaci per il cromosoma Y: • L’assenza della ricombinazione fa si che non esista una mappa genetica per “ancorare” le sequenze. • Estese duplicazioni segmentali con omologia > 99. 95% non permettono di ottenere delle mappe fisiche univoche • Le stesse duplicazioni segmentali creano problemi per l’assemblaggio delle sequenze tramite ricerca di omologia Necessità di adottare strategie di sequenziamento alternative
Cromosoma Y: difficoltà nel sequenziamento SOLUZIONE: SHIMS (single haplotype iterative mapping and sequencing) §Cloni BAC (inserti lunghi) §Genoteca da un unico individuo (per eliminare la diversità allelica): la diversità allelica può essere maggiore della diversità tra paraloghe §Si sfruttano le poche varianti di sequenze paraloghe (PSV) che esistono tra le sequenze duplicate per allineare correttamente le sequenze Palindrome P 3
Il cromosoma X (sequenza completa: 2005) ü Bassa densità genica (1100 geni, 7 geni/Mb): ridotto numero di geni nella coppia autosomica proto-sex oppure, più verosimilmente, pressione selettiva per il trasferimento di geni su autosomi? ü Elevato grado di conservazione inter-specie nell’ordine e contenuto dei geni in confronto ad altri cromosomi di euteri (vedi focus) ü Arricchimento (circa 100) in geni codificanti per antigeni CT (cancer/testis), famiglia di geni espressi normalmente nei testicoli, ma espressi anche in diversi tipi di tumori (vedi focus) üBasso livello di variabilità genetica (a causa del ridotto numero di cromosomi X nella popolazione in confronto al numero di singoli autosomi, 3/4) ü Particolarmente arricchito in elementi LINE L 1 (30% vs. 17% autosomi), soprattuto nella XCR, probabilmente in relazione al meccanismo di compensazione del dosaggio. ü Arricchito in palindromi rispetto agli autosomi ü Elevato numero di malattie X-linked note, dovute alla condizione di emizigosi nei maschi
Il cromosoma X: Focus su conservazione evolutiva (1) Secondo l’ipotesi di Ohno (1967) il cromosoma X dei mammiferi dovrebbe essere evolutivamente conservato quando si confrontino specie differenti. Mouse La conservazione dovrebbe essere la conseguenza del meccanismo di compensazione del dosaggio nei mammiferi, nei quali solo uno dei cromosomi X nelle cellule somatiche della femmina è attivo da un punto di vista funzionale. Traslocazioni (anche reciproche e quindi «bilanciate» ) che sono alla base dei «riassortimenti» citogenetici osservati confrontando autosomi di specie diverse sarebbero mal tollerate per il cromosoma X. Questo perché porterebbero ad uno sbilanciamento funzionale (la parte di X traslocata non viene compensata – la compensazione avviene in cis - mentre la porzione autosomica traslocata potrebbe essere in parte silenziata). L’ipotesi di Ohno è stata verificata in un gran numero di specie di mammiferi, dapprima a livello citogenetico, quindi a livello di sequenza degli interi cromosomi, MA…
Il cromosoma X: Focus su conservazione evolutiva (2) T La maggior parte dei geni segue la legge di Ohno: 656/800 coding genes X-linked umani ha un ortologo del topo, ma ci sono numerosi geni specie-specifici I geni X specifici sono in gran parte «nuovi» (uomo: 76/144) I geni «nuovi» (independently acquired) si trovano in gran parte in regioni ampliconiche o multicopia I geni «single copy» sono quasi tutti conservati I geni «nuovi» sono espressi principalmente nel maschio nelle cellule della linea germinale
Il cromosoma X: Focus su geni cancer/testis. I geni C/T sono raggruppati in numerose famiglie (GAGE, MAGE, ecc) tutte accomunate da: 1) Espressione «fisiologica» limitata ai testicoli 2) Espressione in tumori di differenti organi/tessuti (importanti come target immunoterapici) 3) Forte pressione selettiva per diversificazione e aumento di numero (in particolare quelli sul cromosoma X: CT-X genes). I geni CT-X sono tra i geni del genoma umano caratterizzati da una maggiore velocità di evoluzione. Circa metà dei geni CT si trovano sul cromosoma X, dove rappresentano circa il 10% del cromosoma (circa 100/1000 geni), in gran parte nelle regioni X ampliconiche L’abbondanza di geni CT sul cromosoma X ha un significato evolutivo? Punto su cui riflettere: efficacia maggiore della selezione positiva per alleli recessivi localizzati sul cromosoma X rispetto ad alleli recessivi localizzati sugli autosomi (emizigosi nei maschi). Se, come sembra, l’aumento in geni e la rapida diversificazione degli stessi è vantaggiosa nei maschi, la maggiore efficacia della selezione potrebbe spiegare l’accumulo di geni C/T sul cromosoma X.
Ereditarietà X-linked dominante, penetranza completa - Maschi affetti hanno figlie affette e figli maschi non affetti -Femmine affette trasmettono il carattere (con probabilità 50%) - A livello di popolazione, le femmine sono affette più frequentemente dei maschi (circa il doppio per malattie rare). X-linked recessiva, penetranza completa - Nei pedigree e nella popolazione, manifestano il carattere quasi esclusivamente i maschi (se il carattere è raro) - I pedigree mostrano frequentemente un salto generazionale nella comparsa del carattere - Le femmine sono affette se il padre è affetto e la madre è portatrice NB: Non teniamo conto per ora di varie possibili complicazioni (inattivazione casuale dell’X, penetranza incompleta, espressività variabile, mutazioni de novo, premutazioni, anticipazione, effetti della letalità maschile, ecc. )
Le regioni pseudoautosomiche Due regioni pseudoautosomiche (PAR 1 e PAR 2) all’estremità dei bracci corto e lungo dei due cromosomi sessuali umani, con omologia di sequenza X-Y = 100%. Non sono soggette a compensazione di dosaggio. PAR 1: Xp e Yp; 2. 6 Mb, 24 geni codificanti identici tra X e Y. E’ sede di una ricombinazione obbligata tra X e Y nella meiosi maschile al fine di garantire la corretta segregazione dei cromosomi sessuali (elevato tasso di ricombinazione). Originatasi dopo la divergenza metateri-euteri come traslocazione autosomi-proto-X (residuo della «X-added region» , XAR). Molto differente in diversi mammiferi, sia per contenuto in geni, sia per omologia di sequenza quando esistono regioni ortologhe identificabili (probabilmente a causa dell’effetto mutageno dell’elevato tasso ricombinazione). PAR 2: Xq e Yq; 0. 33 Mb, 5 geni codificanti identici tra X e Y. Ricombinazione non obbligata. Origine recente dopo radiazione uomo-chimp. Compensazione del dosaggio sono nei geni prossimali (inattivati anche nell’Y)
DNA MITOCONDRIALE (mt. DNA) 3. 1 1, 2% (coding)
DNA MITOCONDRIALE
DNA MITOCONDRIALE ED EREDITARIETA’ MATERNA PNAS dicembre 2018
DNA MITOCONDRIALE (mt. DNA) Eteroplasmia: presenza di molecole di mt. DNA differenti in un individuo. L’eteroplasmia può essere limitata ed essere osservata a livello di singolo mitocondrio, cellula, tessuto. Il livello di eteroplasmia (proporzione di DNA mutato rispetto al DNA totale) può variare in tessuti/cellule differenti dello stesso individuo. La variabilità nel grado di eteroplasmia che si osserva in un individuo (e/o in momenti diversi della vita di un individuo) è conseguenza di (1) segregazione casuale del mt. DNA nelle cellule figlie (vegetative segregation in figura), (2) replicazione / degradazione diseguale di molecole di mt. DNA, anche in cellule che non sono in divisione (relaxed replication in figura). Inoltre (3) la selezione può favorire/sfavorire determinate mutazioni Omoplasmia: tutte le molecole di mt. DNA (ad un dato livello di osservazione) sono uguali. Mutazione omoplasmica: mutazione che è presente in tutte le cellule di un individuo (oppure, se specificato, di un certo tessuto…).
DNA MITOCONDRIALE (mt. DNA) Il livello di eteroplasmia è inoltre differente tra cellule della linea germinale femminile e quindi quando si confrontino differenti figli di una femmina eteroplasmica (figura). La variabilità nel grado di eteroplasmia tra figli di una donna eteroplasmica si ritiene sia conseguenza principalmente di un fenomeno di «bottleneck» (riduzione del numero di molecole di mt. DNA) e successiva amplificazione del numero di molecole di mt. DNA che si verificano nella linea germinale materna. Come conseguenza di oscillazioni casuali (accentuate dal piccolo numero di mt. DNA), oociti differenti potrebbero avere proporzioni differenti di mt. DNA mutati. Un oocita maturo ha circa 200 mila mt. DNA (in conseguenza dell’amplificazione), mentre il numero di mt. DNA delle cellule primordiali germinali potrebbe essere di poche decine (in conseguenza del bottleneck).
DNA MITOCONDRIALE (mt. DNA) Variazione del numero mitocondri e del mt. DNA nel lifetime e in diversi tessuti PGC = Primordial Germ Cells ICM = Inner Cell Mass ESC = Embrionic Stem Cells
Malattie mitocondriali primarie Si conoscono mutazioni patogene per tutti i geni del mt. DNA (tabella). La maggior parte delle mutazioni che portano a malattie mitocondriali sono in condizione di eteroplasmia, ma alcune possono essere anche in condizione di omoplasmia (es. LHON) Schon et al. 2012 Nat Rev Genet 13: 878
Mutazioni del mt. DNA coinvolte in malattie multigeniche comuni Si ritiene che mutazioni del mt. DNA possano essere coinvolte nella comparsa di alcune malattie multifattoriali poligeniche come il Morbo di Parkinson. L’accumulo progressivo di mutazioni del mt. DNA nelle cellule somatiche ha anche un ruolo nell’invecchiamento e nella progressione dei tumori
Mutazioni patogene del mt. DNA Effetto soglia: la maggior parte delle mutazioni che portano a malattie mitocondriali sono in condizione di eteroplasmia, ed il fenotipo clinico diviene evidente solo superata una certa proporzione di mitocondri che portano la mutazione. La proporzione di molecole di mt. DNA mutate prende il nome di “carico mutazionale mitocondriale”. Con una sola eccezione nota, questa proporzione deve superare una soglia del 50% o più spesso del 70%, per manifestarsi clinicamente (una sorta di “recessività” non mendeliana). L’esatta proporzione dipende dalla mutazione in questione, dal gene interessato, dal tipo di tessuto interessato, da fattori ambientali e altri fattori genetici (sia mitocondriali che nucleari). L’espressività variabile di alcune malattie del DNA mitocondriale è almeno in parte legata alla proporzione di molecole mutate in soggetti eteroplasmici. Il fenomeno per il quale mutazioni diverse nello stesso gene portano a fenotipi clinici differenti (eterogeneità clinica) è molto diffuso nel caso delle patologie mitocondriali primarie (vedi slide successiva). In alcuni casi addirittura si osserva eterogeneità clinica per una singola mutazione, a seconda del carico genetico. Un esempio è la mutazione 8993 T→G nel gene ATP synthase 6 (ATP 6): con un carico mutazionale > 90%, i pazienti mostrano la “maternally inherited Leigh’s syndrome” (MILS), una encefalopatia letale, mentre un carico mutazionale compreso nel range 70 -90% conduce a pazienti con “neuropathy, ataxia and retinitis pigmentosa” (NARP), una malattia debilitante ad insorgenza tardiva e non fatale.
Mutazioni patogene del mt. DNA: mutazioni in t. RNALeu(UUR) Mutazioni nel gene per mt-t. RNA per la leucina mostrano: - Eterogeneità allelica: la stessa malattia può essere dovuta a mutazioni diverse (alleli) del gene. MELAS dovuta a 5 differenti mutazioni. - Eterogeneità clinica: Mutazioni nello stesso locus danno luogo a patologie differenti: miopatia mit. (MM), encefalomiopatia mit. con acidosi lattica e episodi ictus-like (MELAS), encefalomiopatia mit. (PEM), epilessia mioclonica con fibre rosse sfilacciate (MERRF), miopatia e cardiomiopatia mitocondriale (MMC), DMDF -Eterogeneità clinica a livello di singola mutazione: mutazione np 3243 risulta in MELAS ma può anche risultare in diabete mellito+sordità (DMDF); mutazione in posizione 3256 può dare MELAS o MERRF.
Neuropatia ottica ereditaria di Leber (LHON) La più comune patologia mitocondriale primaria e presenta eterogeneità genica. Appannamento della vista seguito da perdita della vista bilaterale totale o quasi totale in pochi mesi. Dovuta a degenerazione del nervo ottico e dei neuroni della retina. Età d’insorgenza media intorno ai 25 anni, ma può verificarsi anche nei bambini e oltre i 70 anni. La penetranza è incompleta, con un marcato sex bias: la LHON è 5 volte più comune nei maschi che nelle femmine. I geni mutati sono tutti codificanti, e nella maggior parte dei casi sono geni del complesso I (ND). Si conoscono alcune decine di mutazioni, ma tre mutazioni rendono conto del 95% del totale dei casi. Il livello soglia per la manifestazione del fenotipo è pari al 70% nelle famiglie eteroplasmiche, ma sono frequenti anche casi di omoplasmia.
Ereditarietà matrilineare ed eteroplasmia del mt. DNA Il fenotipo è presente sia nei maschi che nelle femmine, ma viene trasmesso solo da queste ultime. Nell’ereditarietà matrilineare “pura”, tutti i discendenti di una femmina affetta saranno affetti, mentre nessuno dei figli di un maschio affetto sarà affetto. A causa della eteroplasmia ed eventi di bottleneck soltanto una parte dei figli di femmine affette sarà in realtà affetto (vedi fratria II, 2 -7; III, 7 -12; IV, 1 -4). Femmine eteroplasmiche non affette (basso carico mutazionale) possono avere figli affetti (alto carico mutazionale). Nel pedigree a fianco questa eventualità non è contemplata. In pedigree di piccole dimensioni si può facilmente confondere con ereditarietà autosomica dominante.