BLAST THE BASIC LOCAL ALIGNMENT AND SEARCH TOOL

BLAST THE BASIC LOCAL ALIGNMENT AND SEARCH TOOL LESSON 4


BLAST • The program compares nucleotide or protein sequences to sequence databases and calculates the statistical significance of matches. • BLAST can be used to infer functional and evolutionary relationships between sequences as well as help identify members of gene families.

• • • http: //blast. ncbi. nlm. nih. gov http: //www. uniprot. org/? tab=blast http: //web. expasy. org/blast/ BLAST “The most Popular Data-Mining Tool ever” “You can do almost everything with BLAST!” üIdentifying orthologs and paralogs üPredicting a protein function üPredicting a protein 3 -D structure üFinding genes in genome üFinding protein family members

DNA can potentially encode 6 different proteins. Bioinformatics and Functional Genomics, 2 nd Edition Jonathan Pevsner John Wiley & Sons, Inc.

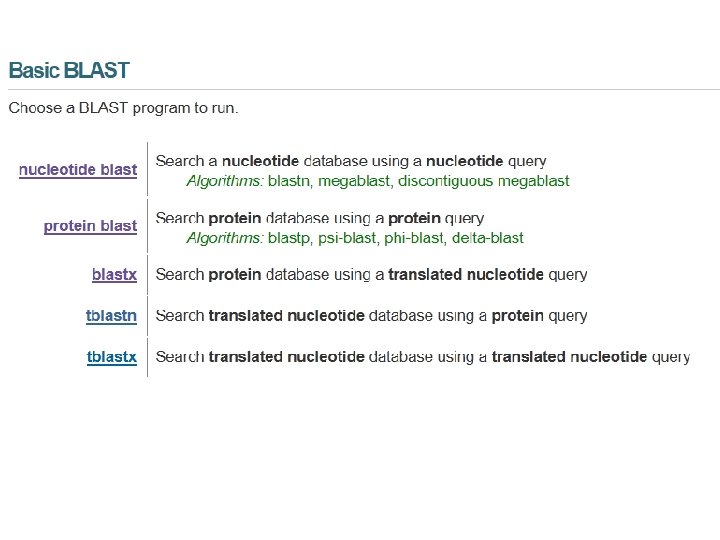
Basic BLAST program Program Input blastn DNA blastp protein blastx DNA tblastn protein tblastx DNA Bioinformatics and Functional Genomics, 2 nd Edition Jonathan Pevsner 1 1 6 6 36 Database DNA protein DNA John Wiley & Sons, Inc.

BLASTING PROTEIN SEQUENCES o protein blast o blastp, psi-blast, phi-blast, delta-blast o Compares a protein sequence with a protein database “find out something about the function of my protein” o tblastn o Compares a protein sequence with a translated nucleotide database “discover new genes encoding simple proteins”
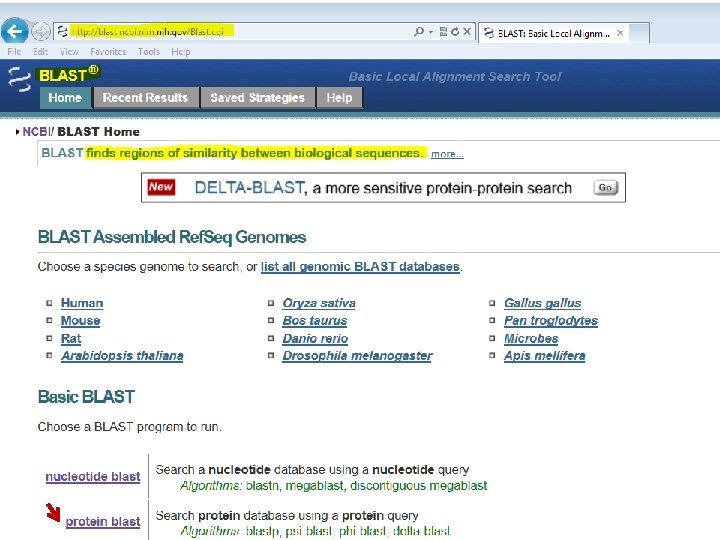

1. 2. 3. 4. 5.

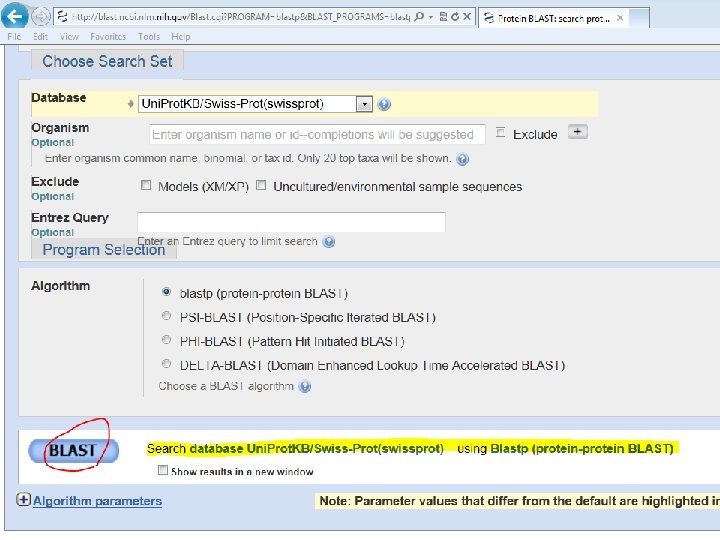
P 09405 • Copy & paste • Enter accession number • Upload file or or

P 09405
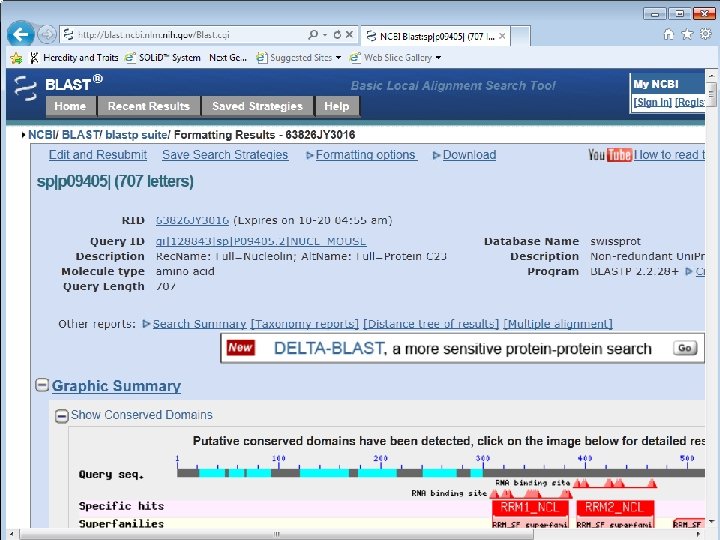

OVERVIEW OF THE BLAST OUTPUT 1. Graphical display 2. Hit list 3. Alignments

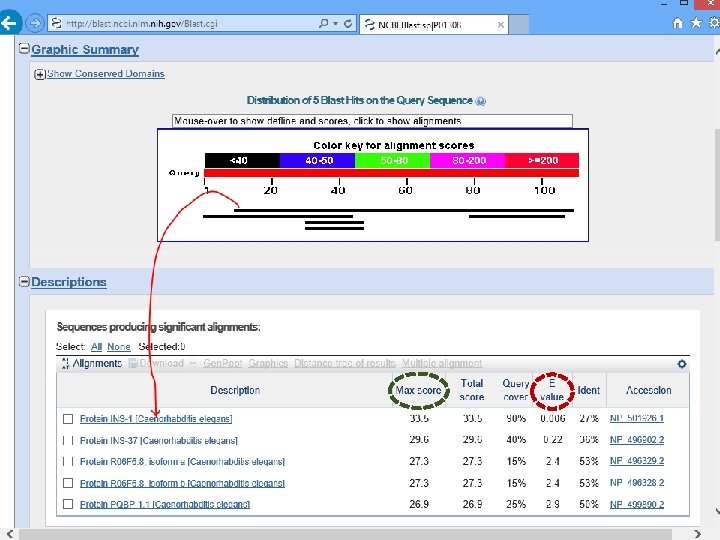
1. Graphic display Each bar represents the portion of another sequence that’s similar to your query.

Useful tool to discover domains

2. Hit List d Ranke ity ilar m i s y b

3. Alignments My sequence Matching sequence Database sequence

o Identities o Positives: Positives fraction of residues that are either identical or similar My sequence Matching sequence Database sequence

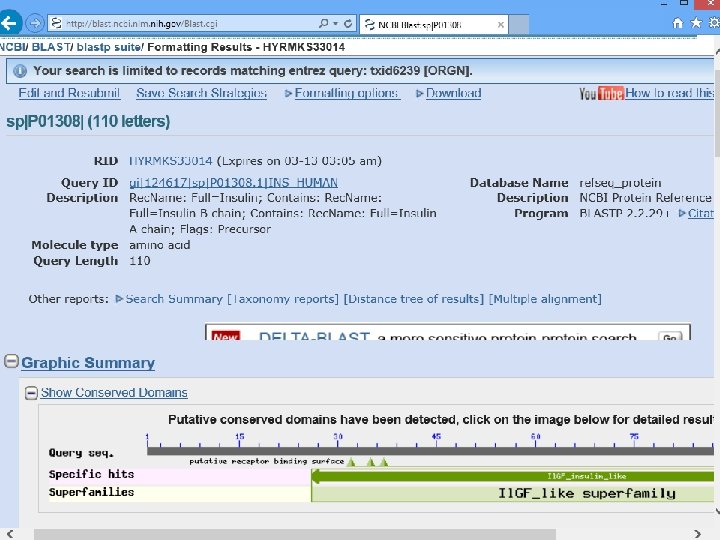
Does C. elegans have INSULIN? Search human insulin against worm Ref. Seq proteins by blastp

sulin n I n a Hum
 / ln 2 Raw Score")
S´= bit score = (l. S - ln. K) / ln 2 Raw Score Bit Score (normalized score) (calculated from a substitution matrix) Bit scores allow you to compare results between different database searches, even using different scoring matrices.
-VALUE • The number of alignments that would be expected by chance alone in")
E(EXPECTATION)-VALUE • The number of alignments that would be expected by chance alone in searching a complete database • Statistical significance

a score of 33. 5 bits or better is expected to occur by chance 0. 6 in 100 times a score of 29. 6 bits or better is expected to occur by chance 22 in 100 times
-VALUE • The smaller the E-value, the more similar the sequences. E = mn")
E(EXPECTATION)-VALUE • The smaller the E-value, the more similar the sequences. E = mn 2 –S´ E (the expect value) = the number of high-scoring segment pairs (HSPs) expected to occur with a score of at least S´ S´ = bit score m, n = the length of two sequences • E decreases with increasing S´. ØVery high scores correspond to very low E values. (Higher scores correspond to better alignments. )
-VALUE • An E value is related to a probability (p). p = 1")
E(EXPECTATION)-VALUE • An E value is related to a probability (p). p = 1 - e-E E 10 5 2 1 0. 05 0. 001 0. 0001 p 0. 99995460 0. 99326205 0. 86466472 0. 63212056 0. 09516258 (about 0. 1) 0. 04877058 (about 0. 05) 0. 00099950 (about 0. 001) 0. 0001000 • Very small E values are very similar to p values. • E values of 0. 05 are statistically significant. Bioinformatics and Functional Genomics, 2 nd Edition Jonathan Pevsner John Wiley & Sons, Inc.

BLAST ALGORITHM How the original BLAST algorithm works
 Bioinformatics and Functional Genomics, 2 nd Edition Jonathan Pevsner John Wiley &")
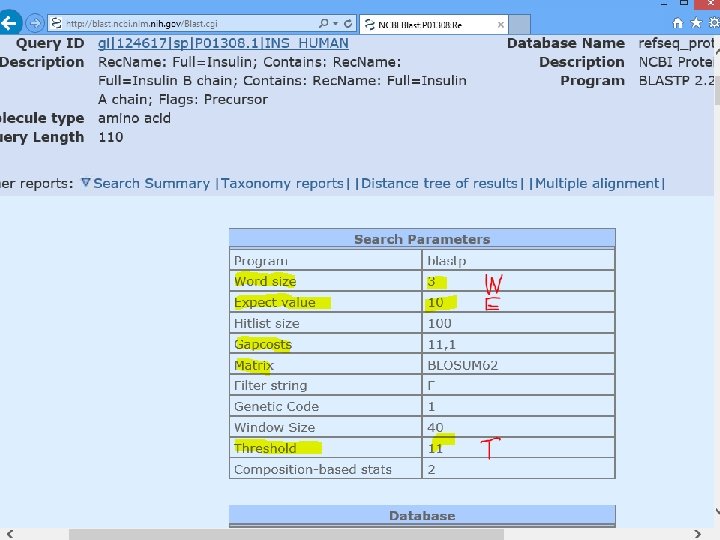
(T =11) Bioinformatics and Functional Genomics, 2 nd Edition Jonathan Pevsner John Wiley & Sons, Inc.

Phase 2: Select all the words above threshold T & Scan for entries that match the compiled list. Phase 3: Extend the database the hits in either direction. Stop when the score drops. Bioinformatics and Functional Genomics, 2 nd Edition Jonathan Pevsner John Wiley & Sons, Inc.

How a BLAST search works “The central idea of the BLAST algorithm is to confine attention to segment pairs that contain a word pair of length W with a score of at least T. ” Altschul et al. (1990) Bioinformatics and Functional Genomics, 2 nd Edition Jonathan Pevsner John Wiley & Sons, Inc.

CONTROLLING BLAST Choosing the right parameters

protein blast ch matches are su 0 1 t a th s n a (10) me The default value chance. y b ly re e m d n u fo e expected to b Lower EXPECT thresholds are more stringent, leading to fewer chance matches being reported. The length of the seed th at initiates an alignment. Lowering the word size yields a more accurate but slower search.

Nucleotide blast

Nucleotide blast er matches w fe s e iv g 8 2 = W = 11. W n a th r te s fa is t bu Megablast is VERY fast for finding closely related DNA sequences! p No cost for opening a ga
")
LOW-COMPLEXITY REGIONS o Regions that contain many identical residues • Runs of proline (P) or acidic amino acids (D, E) • Complicate homology search • Better to exclude – filter!

MASK LOWER CASE LETTERS


HOMEWORK 3 Perform a blastp search at NCBI using the following query of just 12 amino acids: PNLHGLFGRKTG. By default, the parameters are adjusted for short queries. Inspect the search summary of the output. • • What is the E value cutoff? What is the word size? What is the scoring matrix? How do these settings compare to the default parameters?
- Slides: 44