Introduction la Phylognie I Introduction Phylognie HYPOTHSE DE



. Basée sur")



 connectés")




 � M")


 Comment choisir une région de l’ADN qui « reflète")
 de n feuilles:")
 de n feuilles: n-2")
 de n feuilles: n-2")










 �Modèle markovien de substitution le plus simple.")
 �Transitions et transversions ont des taux différents. �Transitions: A")











")








 �Algorithme glouton qui choisit à chaque étape une")



, D(i, j):")




 �On ne compte pas juste le nombre de substitutions,")
 Sk(a) = minb (Si(b) + S(a, b))+ minc (Sj(c)+S(a,")
")
 �Pour retrouver les nucléotides aux nœuds internes, garder des")





- Slides: 71
Introduction à la Phylogénie
I. Introduction - Phylogénie � HYPOTHÈSE DE BASE: Tous les êtres vivants descendent d’un ancêtre commun. Sur une période d’au moins 3. 8 milliards d’années le premier être vivant sur terre n’a cessé de se séparer en espèces différentes. � Les êtres vivants évoluent à partir d’un ancêtre commun par une suite de mutations suivies de spéciations. Tout au long de l’évolution, les gènes accumulent des mutations. Lorsqu’elle sont neutres ou bénéfiques à l’organisme elles sont transmises d’une génération à
Phylogénie �L’isolement d’une population et l’adaptation à son environnement peut entrainer la création d’une nouvelle espèces http: //www. tutorvista. com/content/biology/b iology-iv/biotic-community/speciation. php
Phylogénie � Étude des relations d’évolution entre des groupes d’organismes (espèces, populations). Basée sur la notion d’ « héritage» � Taxonomie: Science qui consiste à classifier identifier et nommer les organismes. Basée sur des caractéristiques communes, différentes du reste de la diversité biologique. Domain, Kingdom, Phylum,
Arbre de Phylogénie The Tree of Life image that appeared in Darwin's On the Origin of Species by Natural Selection, 1859. It was the book's only illustration http: //commons. wikimedia. org/wiki/File: Dar wins_tree_of_life_1859. gif
Arbre de Phylogénie �Premier objectif des études phylogénétiques: Reconstruire l’arbre de vie de toutes les espèces vivantes à partir des données génétiques observées. NASA: http: //www. nasa. g ov
Arbre de Phylogénie �Les arbres de phylogénie sont également utilisés pour représenter l’évolution commune d’une famille de gènes, ou de virus comme le HIV ou l’influenza. Observation de corrélations entre les mutations du gène Myosin avec certains changements anatomiques dans la lignée humaine. MYH 16 chez l’humain très divergeant des autres copies du gène. http: //bio. nyk. ch/Myosi n
II. Définitions formelles � Arbre: Graphe connexe acyclique; Ensemble de nœuds (ou sommets) connectés par des arêtes (ou branches) de telle sorte que toute paire de nœuds est reliée par exactement un chemin. � Arbre raciné: Un nœud est désigné comme étant la racine; permet d’orienter la lecture de l’arbre; le temps s’écoule de la racine vers les feuilles. C H G O M H C G O M
Définitions formelles � Arbre raciné binaire: Chaque nœud interne a deux fils. H C G O M Dans le cas d’un arbre d’espèces : � Les feuilles représentent les espèces (ou séquences) actuelles � La racine représente l’ancêtre commun � Les nœuds internes représentent les événements de spéciation.
Définitions formelles �Un arbre phylogénétique peut-être binaire ou non- binaire. �Un nœud non-binaire représente généralement un nœud non-résolu de l’arbre NJ tree (with weighting) of 119 Bacteria. Asterisks denote anomalously positioned taxa. http: //www. ncbi. nlm. nih. gov/pm c/articles/PMC 540256/figure/fig 3/
Définition formelle �Les nœuds ou arêtes d’un arbre de phylogénie peuvent être étiquetés. Les étiquettes représentent généralement le taux de mutations survenu, ou la date de spéciation R. V. Samonte & Evan E. Eichler Nature Reviews Genetics 3, 65 -72 (January 2002)
Marsupiaux Monotrème s Euthérie ns Monophylie/Paraphylie/Polyphylie Mammifère s http: //du-cote-de-chez-elysiachlorotica. blogspot. ca/2012_10_01_archive. html Tétrapodes: animal du sous-embranchement des vertébrés dont le squelette comporte deux paires de membres et dont la respiration est
Monophylie/Paraphylie/Polyphylie T: arbre raciné. Soit M un groupe d’espèces (actuelles et ancestrales) � M Groupe Monophylétique si le LCA e de M, ainsi que tous ses descendants sont dans M. Autrement dit, M détermine un sous-arbre de T. Exemple dans l’arbre des tétrapodes: Mammifères � M Groupe Paraphylétique si le LCA e de M est dans M, mais que M n’est pas complet, i. e. n’inclue pas toutes les espèces du sous-arbres de racine e. Les Reptiles � M Groupe Polyphylétique si le LCA de M n’est pas dans M. Les tétrapodes à sans chaud ou héméothermes (Mammifères et oiseaux). L’ancêtre des amniotes n’était pas héméotherme.
III. Les caractères ou marqueurs utilisés �Une région spécifique de l’ADN, �Une protéine �Un caractère morphologique �L’ordre des gènes dans le génome �… Les caractères choisis doivent être homologues Hypothèse généralement considérée: Chaque caractère évolue indépendamment des autres.
Les caractères ou marqueurs utilisés �Caractères les plus utilisés pour les études d’évolution: Séquences de nucléotides ou d’AA. �Séquences orthologues dans les espèces étudiées �Effectuer un alignement multiple des séquences �Les caractères représentés par les colonnes de dolphin l’alignement et les états du caractère sont les nucléotides (ou AA observés) ATGACCAACATCCGAAAAACACACCCTCTAATAAAAATCCTC giant sperm whale ATGACCAACATCCGAAAATCACACCCATTAATAAAAATCATT bowhead whale ATGACCAACATCCGAAAAACACACCCACTAATAAAAATTATT right whale ATGACCAACATCCGAAAAACACACCCAGTAATAAAAATTATT minke whale
Choix de marqueurs (séq. d’ADN) Comment choisir une région de l’ADN qui « reflète » l’évolution de tout le génome? Caractéristiques gagnantes: � Marqueur « non-recombinant » . Pour éviter ce problème, choisir des marqueurs uni-parentaux, comme les seq. de mitochondries et de chloroplastes: transmission par la mère uniquement. � Marqueur en copie unique, pour éviter de choisir de mauvais « paralogues » ou: � Marqueurs en copie multiples subissant une « évolution concertée » permettant d’uniformiser toutes les copies. � ARNr: Marqueurs très utilisés pour les études phylogénétiques: �Régions répétées de l’ADN subissant une évolution concertée �Parmi les familles de gènes les plus conservées dans la
IV. L’arbre caché dans la forêt � Arbre non raciné (binaire) de n feuilles: n-2 nœuds internes, n-3 branches internes, et 2 n-3 branches. Chaque branche définit une bipartition de l’ensemble des feuilles. Arbre définit par n-3 bipartitions non-triviales. n=2: 1 2 • Arbre non raciné unique • Arbre raciné unique
L’arbre caché dans la forêt � Arbre non raciné (binaire) de n feuilles: n-2 nœuds internes, n-3 branches internes, et 2 n-3 branches. Chaque branche définit une bipartition de l’ensemble des feuilles. Arbre définit par n-3 bipartitions non-triviales. 3 n=3: 1 2 1 1 3 2 2 • Arbre non raciné unique • 3 arbres racinés 1 3 2 3
L’arbre caché dans la forêt � Arbre non raciné (binaire) de n feuilles: n-2 nœuds internes, n-3 branches internes, et 2 n-3 branches. Chaque branche définit une bipartition de l’ensemble des feuilles. Arbre définit par n-3 bipartitions non-triviales. n=4: 2 4 1 3 3 4 1 2 4 3 1 2 • 3 Arbre non racinés • 15 arbres racinés 1 2 1 1 2 3 2 4 3 4 2 1 3 4 4 3 1 2 4 3
L’arbre caché dans la forêt � Donc le problème d’inférence d’arbres se pose à partir de 3 feuilles pour les arbres racinés, et de 4 feuilles pour les arbres non-racinés. � Cavalli-Sforza et Edwars (1967) ont montré que le nombre Br d’arbres racinés à n feuille est: Br = (2 n-3)!/ 2 n-2 (n-2)! � Le nombre Bu d’arbres non racinés à n feuilles est égal au nombre d’arbres racinés à n-1 feuilles, donc: Bu = (2 n-5)!/ 2 n-3 (n-3)! � Le nombre d’arbres augmente très rapidement avec le nbre de feuilles: Pour n=10, il existe plus de 34 millions d’arbres racinés possibles. Un seul représente la réalité!!
Enracinement �La plupart des méthodes de reconstruction phylogénétiques produisent des arbres non racinés. �Pour un arbre non raciné de n feuilles, 2 n-3 enracinements possibles. Plusieurs méthodes existent: �Enracinement au barycentre: positionner la racine au milieu du chemin séparant les deux feuilles plus éloignées. Hypothèse de l’horloge moléculaire. Applicable uniquement aux arbres valués. �Enracinement en utilisant un « outgroup » . Méthode la plus utilisée. Consiste à rajouter à l’ensemble des séquences des espèces étudiées, une séquence homologue appartenant à une espèce non-
Le kangourou est utilisé comme « outgroup » : Marsupiaux versus mammifères placentaires. http: //cabbagesofdoom. blogspot. ca/2012/06/how-to-root-phylogenetictree. html
Distance topologique �Comment comparer deux arbres T 1, T 2 provenant de données différentes? �Distance la plus utilisée: Robinson-Foulds. Compte le nombre de bipartitions différentes entre T 1 et T 2.
Distance topologique �Distance la plus utilisée: Robinson-Foulds. Compte le nombre de bipartitions différentes entre T 1 et T 2 Bipartitions. non-triviales D D E F C B C A A E F B CD|ABEF EF|ABCD AB|CDEF CD|ABEF EB|ACDFAF|BCDE
Distance topologique �Distance la plus utilisée: Robinson-Foulds. Compte le nombre de bipartitions différentes entre T 1 et T 2 Bipartitions. non-triviales D D E F C B C A A E F B CD|ABEF EF|ABCD AB|CDEF CD|ABEF EB|ACDFAF|BCDE Disance topologique d. T (T 1, T 2)= 4
Distance topologique �Distance la plus utilisée: Robinson-Foulds. Compte le nombre de bipartitions différentes entre T 1 et T 2. �Un arbre non raciné de n feuilles a n-3 branches internes (bi-partitions non-triviales). Donc distance topologique maximale entres deux arbres non racinés est d. M (T 1, T 2)=2(n-3) �Généralement, la distance tolologique est normalisée: RF(T 1, T 2) = d. T(T 1, T 2)/d. M(T 1, T 2)
VI. Modèles d’évolution moléculaire �Distance évolutive d entre deux séquences: nombre moyen de substitutions/site s’étant produites depuis la divergence de ces deux séquences à partir d’un ancêtre commun. �Estimation des distances évolutives à la base de la plupart des méthodes de reconstructions phylogénétiques. �Construction d’une matrice de distance contenant les distance évolutives entre paire de séquences: Première étape des méthodes phylogénétiques.
Divergence observée �Calculée directement à partir de la distance d de Levenshtein ou de Hamming (substitutions) entre deux séquences (ADN ou protéines). �Taux de divergence = d/n où n est la taille des séquences. �Pour deux séquences aléatoires d’ADN, le taux de divergence est égal à 0. 25 �Divergence observée: seule mesure directement accessible. �Pas un bon estimateur à part pour les séquences très proches: tendance à sous-estimer la distance évolutive réelle.
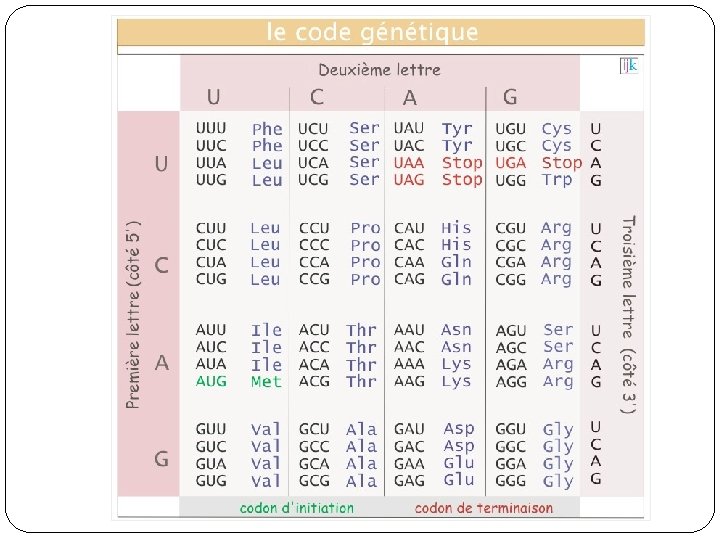
Modèle markovien de l’évolution �Calcul d’une probabilité de transition d’un état à un autre �Calcul d’une matrice 4 x 4: �mij (i ≠j) : taux de substitution instantané de l’état i à l’état j. � 1 -mi : taux de conservation instantané du nucléotide i. �Q: matrice des taux du precessus de Markov. La
Modèle de Jukes et Cantor (JC 69) �Modèle markovien de substitution le plus simple. �Considère le même taux de substitution instantané pour chacun des changements possible, et un seul taux de conservation global. �m/4: taux moyen instantané de substitution.
Modèle de Kimura (K 80) �Transitions et transversions ont des taux différents. �Transitions: A G, C T � Transversions: A T, T G, A C, C G �k rapport entre le taux de transitions et le taux de transversions.
Sélection naturelle �Processus par lequel certaines modifications apparaissant par hasard chez certains individus dans une population sont favorisées et fixées, tandis que d’autres sont défavorisées et perdues. �Concept initialement formulé par Darwin, basée sur une observation des phénotypes, mais la sélection naturelle affecte également le génotype. �Peut mener à la création de nouvelles espèces.
Distance synonyme/non-synonyme pour les séquences codantes Les gènes sont soumis à plusieurs types de sélection naturelle: �Sélection positive: Processus qui encourage la rétention des mutations qui sont bénéfiques pour un individu. �Sélection négative ou purificatrice: Processus qui tend à faire disparaître des mutations nuisibles. �Sélection neutre: Absence de sélection positive ou négative. Dans le cas de séquences qui ne sont affectées par aucune pression sélective. Peuvent être modifiées sans conséquences sur l’organisme.
Distance synonyme/non-synonyme pour les séquences codantes �Basée sur la comparaison des substitutions synonymes et non-synonymes (effet sur les codons) �Substitution non-synonyme (non-silencieuse): substitution provoquant la modification d’un acide aminé. �Substitution synonyme (silencieuse): substitution ne provoquant pas la substitution de l’acide aminé initial. http: //en. wikipedia. org/wiki/Missense_mutation
Distance synonyme/non-synonyme pour les séquences codantes �Sites synonymes et non-synonymes: � 100% des mutations touchant la 2ème base des codons sont non-synonymes �Sous l’hypothèse que les fréquences nucléotidiques sont égales et que les mutations se font au hasard, 95% des mutations touchant la 1ère base et 28% des mutations touchant la 3ème base sont nonsynonymes. �Distances synonymes et non-synonymes: �d. S (aussi notée KS): Distance synonyme � d. N: Distance non-synonyme
Distance synonyme/non-synonyme pour les séquences codantes Identification du type de sélection: �Sélection négative: Déficit de substitutions nonsynonymes attendu d. N/d. S < 1 �Sélection neutre: Aucun déficit en subst. nonsynonymes attendu d. N/d. S ≈ 1 �Sélection positive: Excès de subst. nonsynonymes attendu d. N/d. S > 1
Inférence d’arbres phylogénétiques � Méthodes de distance �Input: Matrice de distances D �Construire un arbre qui « réalise » cette matrice: chaque paire (x, y) de feuilles est reliée par un chemin dont le score est égal à la distance D(x, y) entre x et y. � Méthodes de parsimonie: Arbre qui explique l’évolution des espèces par un nombre minimum de mutations. Deux composantes principales: �Calcul d’un score d’un arbre donné. �Recherche, parmi tous les arbres, l’arbre de score minoimal. � Méthodes probabilistes �Maximisation de la vraisemblance d’un arbre �Inférence Bayésienne, basée sur la probabilité postérieure des hypothèses en fonction des données.
VII. Méthodes de distance �Étant donnée une matrice de distance, existe-t-il un arbre binaire qui « réalise » la matrice? 1 AA A 0 B 5 C 8 D 2 E 12 11 B 5 0 9 13 12 C 8 9 0 6 5 D 12 13 6 0 3 E 11 12 5 3 0 4 3 A B 1 C 3 2 D 1 E
Condition des 4 points � Théorème: Il existe un arbre réalisant la matrice de distance si et seulement si la matrice satisfait la condition des quatre points. � Condition des 4 points: Pour tout choix de 4 feuilles A, B, C, D, deux des sommes suivantes sont égales et supérieures à la 3ème: D(A, B) + D(C, D), D(A, D) + B A D(B, C) et D(A, C)+D(B, D) A B D C A C B D C D
Distances additives �Une distance qui satisfait la condition des 4 points est une distance additive. A A A B C D 0 B C D 3 3 5 0 4 6 0 4 0 C A 1 1 1 B 2 3 D
Distance ultramétrique �Condition des 3 points: Pour tout choix de 3 feuilles A, B, C, parmi les trois distances D(A, B), D(A, C) et D(B, C), deux sont égales et supérieures à la troisième. D(A, C) = D(B, C) D(A, B) A B C
Distance ultramétrique �Condition des 3 points: Pour tout choix de 3 feuilles A, B, C, parmi les trois distances D(A, B), D(A, C) et D(B, C), deux sont égales et supérieures à la troisième. �Une distance qui satisfait la condition des 3 points est dite ultramétrique. �Une distance ultramétrique est une distance D(A, C) = additive. Le contraire n’est pas vrai. D(B, C) D(A, B) A B C
Distance ultramétrique � Une distance ultramétrique satisfait l’inégalité ultratriangulaire: Dik ≤ max (Dij, Djk) pour tous i, j, k Tous les chemins de la racine à n’importe quelle feuille de la même longueur. � Un arbre « associé » à une distance ultramétrique satisfait la théorie de l’horloge moléculaire: taux de mutation constant sur toutes les branches. D(A, C) = D(B, C) D(A, B) A B C
Arbre ultramétrique T est un arbre ultramétrique associé à la distance ultramétrique D ssi: � T contient n feuilles, chacune étiquetée par une ligne de D; � Chaque nœud interne est étiqueté par une case de D et a au moins deux fils; � Le long d’un chemin de la racine à une feuille les valeurs des étiquettes des nœuds décroissent strictement; � Pour deux feuilles quelconques i, j, D(i, j) est l’étiquette du dernier ancêtre commun de i et j dans T. T, s’il existe, est une représentation compacte de D. Remarque: T a au plus n-1 nœuds internes. Donc, si D
Algorithme UPGMA �UPGMA: Algorithme de classification ascendante hiérarchique. �Procède par regroupement des séquences les plus proches. À chaque étape, les deux regroupements les plus « proches » sont fusionnés. �Si D est une distance ultramétrique, alors UPGMA construit l’arbre ultramétrique associé.
Algorithme UPGMA �n séquences; Di, j: Distance entre les séquences i et j. �dij: Distance entre deux regroupements Ci et Cj. Moyenne des distances des paires de séquences entre les deux regroupements. �Si Ck = Ci U Cj et Cl est un autre regroupement, alors:
Distance/arbre ultramétrique Théorème: Si D est une matrice ultramétrique, alors l’arbre ultramétrique de D est unique. Preuve: Dans la construction de l’arbre, les classes sont « forcées » , i. e. ne peuvent pas être déterminées autrement, et les positions de ces classes sont forcées aussi. Conséquence: Si D reflète effectivement la distance d’évolution entre les espèces, alors l’arbre obtenu est nécessairement le vrai arbre. Théorème: Si D est ultramétrique, alors l’arbre ultramétrique peut-être construit en temps O(n 2). De plus, on peut déterminer en O(n 2) si une distance est ultramétrique ou non.
Que signifient des données ultramétriques? �Distances étiquetant les arbres ultramétriques supposées refléter le temps qui s’est écoulé depuis la séparation des deux espèces. �Théorie de l’horloge moléculaire (1960): Pour une protéine donnée, le taux de mutations acceptées par intervalle de temps est constant. �Donc, si k mutations acceptées entre les protéines A et B, on peut estimer à k/2 le nombre de mutations survenues sur chaque branche depuis l’ancêtre commun de A et B. Permet d’obtenir des données ultramétriques.
Distance/arbre additif Soit D une distance pour n séquences. T: Arbre contenant au moins n noeuds (dont les feuilles). Chaque ligne de D correspond à un nœud différent, et les arêtes sont étiquetées. �T arbre additif pour D si pour toute paire de nœuds (i, j), le poids total du chemin de i à j est D(i, j).
Distance/arbre additif �Problème: Trouver un arbre additif pour D ou déterminer qu’un tel arbre n’existe pas. �Théorème: Il existe un arbre additif pour D ssi D est une distance additive (i. e. vérifie la condition des 4 points). �Distance additive: Contrainte moins forte que la contrainte ultramétrique. Une distance ultramétrique est additive. Le contraire n’est pas vrai. �Cependant, les données réelles sont rarement
Neighbor-Joining (Saitou et Nei en 1986) �Algorithme glouton qui choisit à chaque étape une paire de feuilles voisines. �Obtient un arbre additif correspondant à une distance additive. �En général NJ est une approximation du « minimum d’évolution » �Minimum d’évolution: Parmi toutes les topologies d’arbres, choisir celle minimisant la somme des longueurs de branche calculées en utilisant la méthode des moindres carrés, i. e. longeurs de branche minimisant: Q = Si<j wij (Dij-dij)2 wij: pondération associée à chaque couple (i, j).
Neighbor-Joining � Paire de feuilles voisines: Deux feuilles de T ayant le même père. 1 3 4 5 6 2 7 (1, 2), (6, 7), (4, 5), ((1, 2), 3), ((4, 5), (6, 7)), (1, 2), 3)) � Un arbre est déterminé par l’ensemble des (n-2) paires de voisins qu’il contient.
Neighbor-Joining � Choisir deux objets i, j garantis d’être voisins dans un arbre additif. � Supprimer i, j de la liste des objets et rajouter le nœud créé k correspondant au père commun de i et j. � Distance de k à une feuille m quelconque: 3 i 1 4 m 5 6 k j 2 7 D(k, m) =1/2 (D(i, m) + D(j, m)- D(i, j) )
Neighbor-Joining �Comment déterminer, à partir de D, deux feuilles qui spmt nécessairement voisines dans un arbre additif de D? �Il ne suffit pas de choisir une paire d’objets dont la distance 1 est minimal: 2 1 4 3 1 (1, 2) de distance minimale mais pas voisines dans l’a 1 4 4
Neighbor-Joining �L: Ensemble des feuilles d’un arbre additif. �Pour tout (i, j), D(i, j): valeur obtenue en soustrayant de D(i, j) la distance moyenne de i et j à toutes les autres feuilles. D(i, j) = D(i, j) – (ri+rj) Théorème: Si T est un arbre additif pour la distance additive D, si (i, j) est une paire de feuille telle que D(i, j) est minimal parmi toutes les paires de feuilles, alors i et j sont voisines dans T.
VIII. Méthodes de parcimonie �Basées sur le principe de maximum de parcimonie: La meilleure hypothèse pour expliquer un processus est celle qui fait appel au plus petit nombre d’événements. �À la différence des méthodes de distances, considère chaque site d’un alignement multiple individuellement. Sous-entend l’hypothèse d’indépendance des sites. �Méthode générale: �Considérer toutes les topologies d’arbres possibles sur un ensemble de feuilles; �Calculer un poids pour chaque arbre; �Sélectionner un arbre de poids minimal.
Méthodes de parcimonie �Pondération d’un arbre: Affecter des séquences aux nœuds internes de telle sorte à minimiser le poids total de l’arbre (somme des distances des branches). �Exemple: AAA S 1: AAG S 2: AAA S 3: GGA S 4: AGA 1 S 1 AAA S AGA 1 S S A 1 A G A 2 A A G 3 G A A G 4 A Poids de l’arbre: 3 S S S S 1 3 2 4 1 4 2 3
Méthodes de parcimonie �Pondération d’un arbre: Affecter des séquences aux nœuds internes de telle sorte à minimiser le poids total de l’arbre (somme des distances des branches). �Exemple: AAA AAA S 1: AAG S 2: AAA S 3: GGA S 4: AGA 1 1 AAA AAA 1 2 S 1 S 2 S 3 S 4 S S A A G A A A G G A A G A 1 AAG GGA AAA 3 2 4 Poids de l’arbre: 3 4 1 S AAA 1 S 2 S AAG AGA AAA 1 4 2 S GGA 3
Parcimonie pondérée (Algorithme de Sankoff) �On ne compte pas juste le nombre de substitutions, mais un poids S(a; b) pour la substitution de a en b. �Étiqueter les nœuds internes de telle sorte a minimiser le poids total de l'arbre. �Par récurrence: étiquette d'un nœud déduite des étiquettes des nœuds fils. �Sk(a): poids du sous-arbre de racine k, sous la condition que k est étiqueté par a.
Parcimonie pondérée (Algorithme de Sankoff) Sk(a) = minb (Si(b) + S(a, b))+ minc (Sj(c)+S(a, c) k: a i b Sk(a) = 0 k: a Sk(b) = ∞ c j
Parcimonie pondérée (Algorithme de Sankoff)
Parcimonie pondérée (Algorithme de Sankoff) �Pour retrouver les nucléotides aux nœuds internes, garder des pointeurs lk(a) et rk(a) pour chaque a et chaque nœud k, et rajouter les deux instructions suivante dans le bloc de récurrence: Poser lk(a) = argminb(Si(b) + S(a, b)) Poser rk(a) = argminb(Sj(b) + S(a, b)) �Pour retrouver une assignation correcte pour les nœuds internes, choisir un nucléotide à la racine qui donne lieu à un poids S 2 n-1(a) minimal, et suivre les pointeurs. �Complexité: Pour un nœud donné, il faut calculer 2|S|2 minima. D’où, complexité de l’algorithme en O(n|S|2 ) où n est la taille de l'arbre (nombre de nœuds).
T T T, C 5 3 4 5 11 A T C G S(a, b) = 1 si a ≠b 10 4 3 3 4 T A, T, C, G 2 2 T T, C 2 1 1 2 9 TT, G 8 7 2 1 1: C 2: T ∞ ∞ 0 ∞ ∞ 3: G ∞ ∞ ∞ 0 4: T ∞ 0 ∞ ∞ 0 5: A 6: T ∞ ∞ 0 ∞ ∞
Parcimonie traditionnelle Algorithme de Fitch �Minimiser le nombre de substitutions de caractères. Garder à chaque nœud une liste de nucléotides « valides » . C: poids courant de l’arbre.
Parcimonie traditionnelle Algorithme de Fitch �Pour retrouver les nucléotides nœuds internes: Choisir un nucléotide dans R 2 n-1(racine) puis descendre dans l'arbre. Si on a choisit a pour k, alors, pour le fils i de k, choisir a si possible, si non choisir un nucléotide au hasard dans Ri. �Complexité: O(n|S|) �Observation: Le poids minimal d’un arbre calculé par l’algorithme de Fitch est indépenant du choix de la racine. Conséquence: on n’a pas besoin de tester tous les arbres racinés possibles.
T 11 R 11 = {T} C = 0+1 +1 +1 10 T R 10 = {T} T R 9 = {G, T, A} 9 T 7 R 7 = {C, T} 1: C T R 8 = {G, T} 8 2: T 3: G 4: T 5: A 6: T R 1 = {C} R 2 = {T} R 3 = {G} R 4 = {T} R 5 = {A}R 6 = {T}
Parcimonie traditionnelle Algorithme de Fitch �Problème de la parcimonie traditionnelle: Certaines assignations possibles des nœuds internes ne sont jamais considérées.