HOW TO RUN NAMD Protein structure File Tutorial









 Acceptable Values: positive decimal cutoff")


 • Of the four files (pdb, psf, topology")



 and perform the")

- Slides: 19
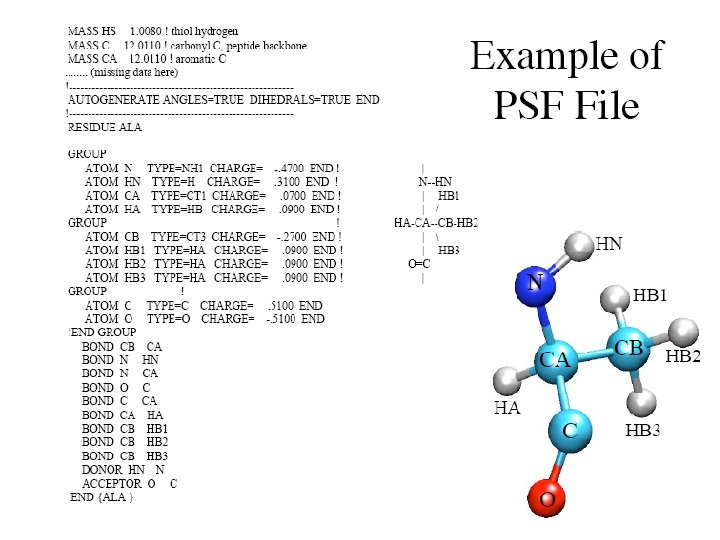
HOW TO RUN NAMD
Protein structure File
Tutorial web page: • http: //www. ks. uiuc. edu/Research/namd/2. 6 b 1/ug/
NAMD configuration parameters ; Timestep parameters ·numsteps number of timesteps > Acceptable Values: positive integer Description: The number of simulation timesteps to be performed. An integer greater than 0 is acceptable. The total amount of simulation time is numsteps x timestep. ·timestep <timestep size (fs) > Acceptable Values: non-negative decimal Default Value: 1. 0 Description: The timestep size to use when integrating each step of the simulation. The value is specified in femtoseconds. ·firsttimestep <starting timestep value > Acceptable Values: non-negative integer Default Value: 0 Description: The number of the first timestep. This value is typically used only when a simulation is a continuation of a previous simulation. In this case, rather than having the timestep restart at 0, a specific timestep number can be specified. ·stepspercycle <timesteps per cycle> Acceptable Values: positive integer Default Value: 20 Description: Number of timesteps in each cycle. Each cycle represents the number of timesteps between atom reassignments. For more details on nonbonded force evaluation, see Section 5. 1.
Simulation space partitioning ·cutoff local interaction distance common to both electrostatic and van der Waals calculations (Å) Acceptable Values: positive decimal Description: See Section 5. 1 for more information. ·switching use switching function Acceptable Values: on or off Default Value: off Description: If switching is specified to be off, then a truncated cutoff is performed. If switching is turned on, then smoothing functions are applied to both the electrostatics and van der Waals forces. For a complete description of the nonbonded force parameters see Section 5. 1. If switching is set to on, then switchdist must also be defined. ·switchdistance at which to activate switching function for electrostatic and van der Waals calculations (Å) Acceptable Values: positive decimal cutoff Description: Distance at which the switching function should begin to take effect. This parameter only has meaning if switching is set to on. The value of switchdist must be less than or equal to the value of cutoff, since the switching function is only applied on the range from switchdist to cutoff. For a complete description of the non-bonded force parameters see Section 5. 1.
·pairlistdistance between pairs for inclusion in pair lists (Å) Acceptable Values: positive decimal cutoff Default Value: cutoff Description: A pair list is generated pairlists. Per. Cycle times each cycle, containing pairs of atoms for which electrostatics and van der Waals interactions will be calculated. This parameter is used when switching is set to on to specify the allowable distance between atoms for inclusion in the pair list. This parameter is equivalent to the X-PLOR parameter CUTNb. If no atom moves more than pairlistdistcutoff during one cycle, then there will be no jump in electrostatic or van der Waals energies when the next pair list is built. Since such a jump is unavoidable when truncation is used, this parameter may only be specified when switching is set to on. If this parameter is not specified and switching is set to on, the value of cutoff is used. A value of at least one greater than cutoff is recommended. ·Etc. .
Simulatinf poly-alanine
# This is a test namd configuration file timestep numsteps structure parameters coordinates exclude 1 -4 scaling outputname margin stepspercycle temperature 1. 0 100000 alanin. psf alanin. params alanin. pdb scaled 1 -4 0. 4 output 1. 0 20 300 langevin on langevin. Damping 5 langevin. Hydrogen langevin. Temp 300 switching switchdist cutoff pairlistdist on 7. 0 8. 0 9. 0 IMDon IMDport IMDfreq IMDwait yes 2030 1 on no
Generating a Protein Structure File (PSF) • Of the four files (pdb, psf, topology and energy parameter files), an initial pdb file will typically be obtained through the Protein Data Bank, • and the parameter and topology files for a given class of molecule may be obtained via the Internet at http: //www. pharmacy. umaryland. edu/faculty/amackere/for ce fields. htm. • The psf file must be created by the user from the initial pdb and topology files.
REMARK REMARK REMARK ATOM ATOM ATOM ATOM ATOM FILENAME="/usr/people/nonella/xplor/benchmark 1/ALANIN. PDB" PARAM 11. PRO ( from PARAM 6 A ) ====== PROTEIN PARAMETERS: PEPTIDE GEOMETRY FROM RAMACHANDRAN ET AL BBA 359: 298 (1974) TORSIONS FROM HAGLER ET AL JACS 98: 4600 (1976) LENNARD-JONES NONBONDED PARAMETERS WITH SPECIAL TREATMENT OF 1: 4 CARBON-CARBON INTERACTIONS: JORGENSON ET. AL. JACS 103: 3976 -3985 WITH 1 -4 RC=1. 80/0. 1 DATE: 16 -Feb-89 11: 21: 32 created by user: nonella 1 CA ACE 1 -2. 184 0. 591 0. 910 1. 00 7. 00 MAIN 2 C ACE 1 -0. 665 0. 627 0. 966 1. 00 0. 00 MAIN 3 O ACE 1 -0. 069 1. 213 1. 868 1. 00 0. 00 MAIN 4 N ALA 2 0. 000 1. 00 3. 00 MAIN 5 H ALA 2 -0. 490 -0. 462 -0. 712 1. 00 0. 00 MAIN 6 CA ALA 2 1. 450 0. 000 1. 00 7. 00 MAIN 7 CB ALA 2 1. 969 -0. 670 -1. 262 1. 00 0. 00 MAIN 8 C ALA 2 2. 010 1. 413 0. 000 1. 00 0. 00 MAIN 9 O ALA 2 2. 911 1. 748 0. 767 1. 00 MAIN 10 N ALA 3 1. 488 2. 280 -0. 863 1. 00 0. 00 MAIN 11 H ALA 3 0. 770 1. 998 -1. 467 1. 00 4. 00 MAIN 12 CA ALA 3 1. 981 3. 643 -0. 909 1. 00 7. 00 MAIN 13 CB ALA 3 1. 147 4. 464 -1. 880 1. 00 0. 00 MAIN 14 C ALA 3 1. 865 4. 326 0. 444 1. 00 0. 00 MAIN 15 O ALA 3 2. 801 4. 963 0. 924 1. 00 0. 00 MAIN 16 N ALA 4 0. 710 4. 211 1. 093 1. 00 9. 00 MAIN 17 H ALA 4 -0. 026 3. 700 0. 697 1. 00 0. 00 MAIN 18 CA ALA 4 0. 541 4. 841 2. 388 1. 00 7. 00 MAIN 19 CB ALA 4 -0. 809 4. 462 2. 976 1. 00 8. 00 MAIN 20 C ALA 4 1. 591 4. 371 3. 381 1. 00 0. 00 MAIN
• • remark - parameter file PARAM 19 remark PEPTIDE GEOMETRY FROM RAMACHANDRAN ET AL BBA 359: 298 (1974) remark TORSIONS FROM HAGLER ET AL JACS 98: 4600 (1976) remark JORGENSEN NONBOND PARAMETERS JACS 103: 3976 -3985 WITH 1 -4 RC=1. 80/0. 1 • • • set echo=false end !! - PEPTIDE GEOMETRY TO GIVE RAMACHANDRAN ET AL BBA 359: 298 (1974) !! - PEPTIDE TORSIONS FROM HAGLER ET AL JACS 98: 4600 (1976) !! - NONBONDED TERMS JORGENSEN JACS 103: 3976 W/ RC 1 -4 = 1. 80 EC 1 -4 = 0. 1 !! The default h-bond exponents are now 6 -repul 4 -attr !! ++++ ATOMTYPE OS (IN METHYL ESTER) ADDED FOR CHARMM COURSE /LN ++++ !! Switched from Slater-Kirkwood to simple mixing rules - AB !! Hbond parameters based on comparisons of dimer results with !! ab initio calculations. - WER 12/19/84 !! Grouping of atom types for VDW parameters - BRB 1/3/85 • • • • bond C bond C bond C bond C C 450. 0 1. 38! B. R. GELIN THESIS AMIDE AND DIPEPTIDES CH 1 E 405. 0 1. 52! EXCEPT WHERE NOTED. CH 1 E, CH 2 E, CH 3 E, AND CT CH 2 E 405. 0 1. 52! ALL TREATED THE SAME. UREY BRADLEY TERMS ADDED CH 3 E 405. 0 1. 52 CR 1 E 450. 0 1. 38 CT 405. 0 1. 53 N 471. 0 1. 33 NC 2 400. 0 1. 33! BOND LENGTH FROM PARMFIX 9 FORCE K APROXIMATE NH 1 471. 0 1. 33 NH 2 471. 0 1. 33 NP 471. 0 1. 33 NR 471. 0 1. 33 O 580. 0 1. 23 OC 580. 0 1. 23! FORCE DECREASE AND LENGTH INCREASE FROM C O OH 1 450. 0 1. 38! FROM PARMFIX 9 (NO VALUE IN GELIN THESIS) OS 292. 0 1. 43! FROM DEP NORMAL MODE FIT
• By using topology file and parameter files you will create alanine. psf file • http: //www. ks. uiuc. edu/Training/Tutorials/ namd/namd-tutorial-win. pdf
• Exercise: repeat the steps in the manual (for ubiquitin) and perform the anlyses in a water box for a temperature between 250 K and 400 K for 2 ns (how many time steps with 2 fs timestep). ASLIHAN AYTUĞ BESRAY GÜNEŞ GÜZİN EMRE MURAT NURCAN ORHAN OSMAN ÖZGE PINAR SEMİH SİNAN ŞERİFE 250 275 300 325 350 375 400 450