Gel filtration Chromatography 1 Desalting group separation separate
: separate 2. the target protein from low-molecular")


 1. Sample pass through a 0. 22 um protein-compatible filter,")
")


 eg. anion exchange gel, binding")



- Slides: 24
Gel filtration Chromatography 1. Desalting (group separation): separate 2. the target protein from low-molecular mass contaminants. 2. Change buffer 3. protein fractionation
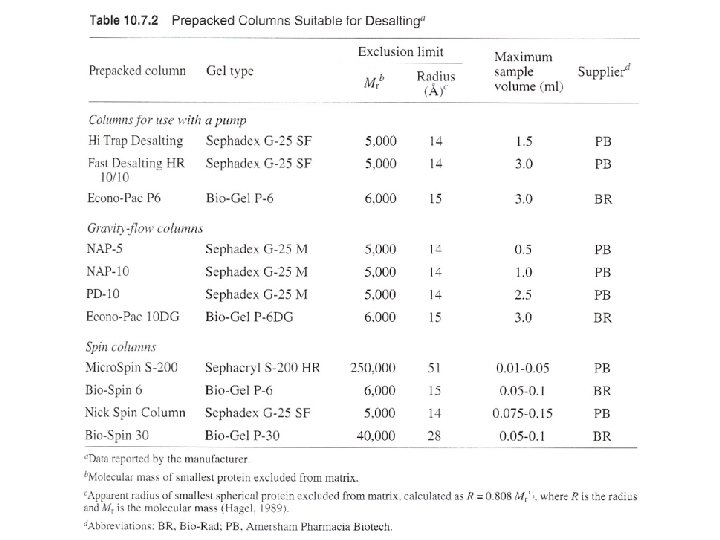
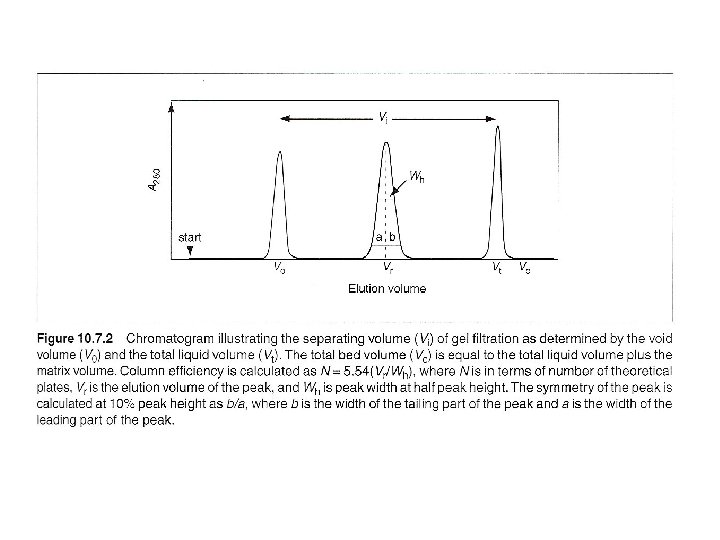
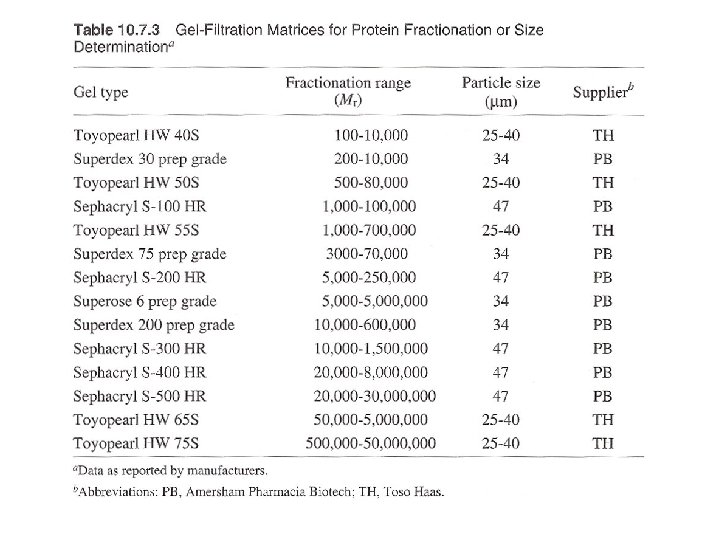
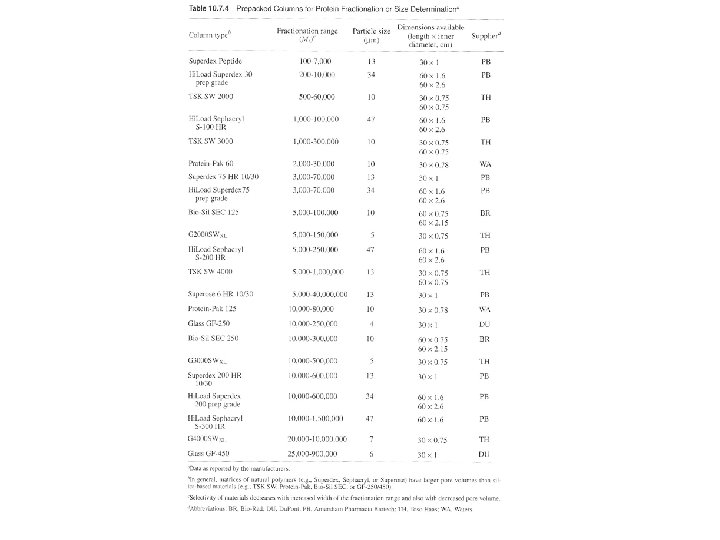
Three ways: columns with pump, gravity-flow columns, spin columns - Many pre-packed columns are available. - Can buy gel matrics, and prepare the gel, and pack the columns (10 -53 to 10 -57, or according to the suppliers’ suggestions). - Use Blue dextran 2000 (blue) or vitamine B 12 (yellow) as void volumn (V 0) marker. - Use acetone (5 mg/ ml) as total volumn (Vt) marker. (by absorbance at OD 280) - Gel matrx volumn + Vt = bed-volumn (Vc) - Vi (separating volumn) = Vt-V 0
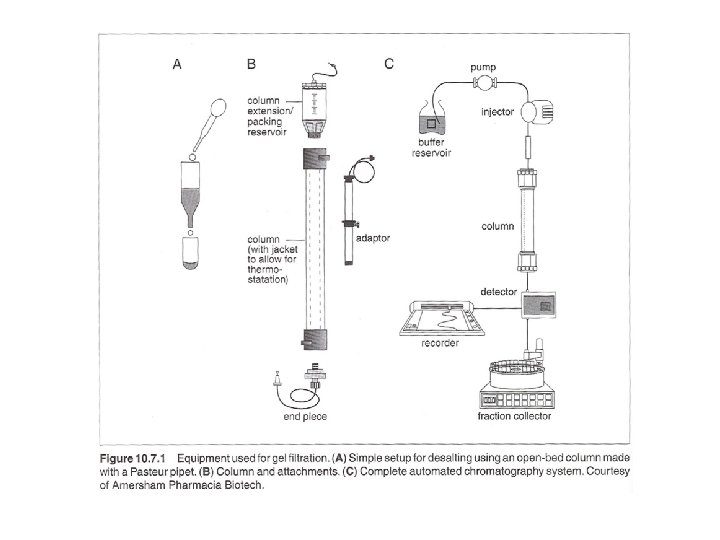
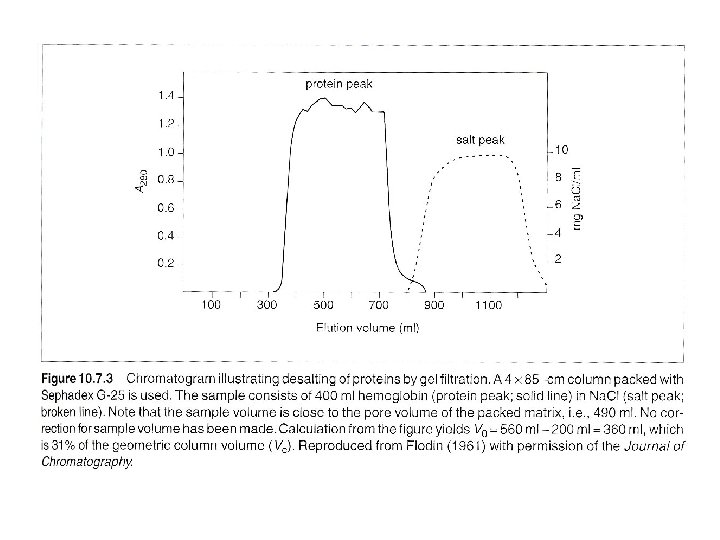
Desalting (or changing buffer) 1. Sample pass through a 0. 22 um protein-compatible filter, and reach the same temperature as column. 2. Run two bed-volumn buffer, and allow the baselines of detector and recorder to stabilize. 3. Load sample, volumn needs to < or = Vi (Or ~ 50% of bad volumn of non-rigid matrics (agarose), ~ 30% of bad volumn of rigid matrics (silica) (To determine Vr (elution volumn): flow rate times time from the start point to the apex of the peak. ) 4. To desalt a protein: first pass buffer of voild volumn reduced by half the applied sample volumn – waste 5. then apply buffer of applied sample volumn – collect. 6. Wash column with > 1 column volumn of buffer with an antibacterial agent and store.
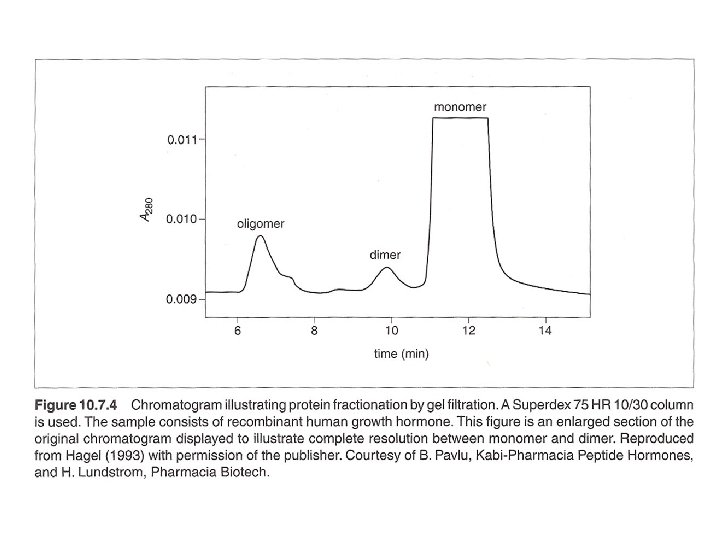
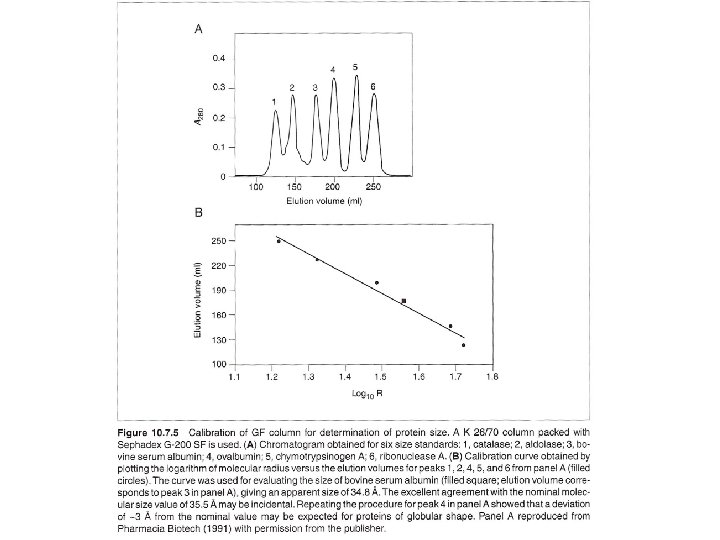
Gel filtration for protein fractionation (usually used in the polishing step in protein purification) - Columns are different from desalting - Sample volumn about 2% of the bed volumn. - Purity of the peak can be checked by HPLC or electrophotresis. - Proteins molecular weight could be determined by Vr (elution volumn). But mass spectrophotometry (MS) can do better job in determination of molecular mass.
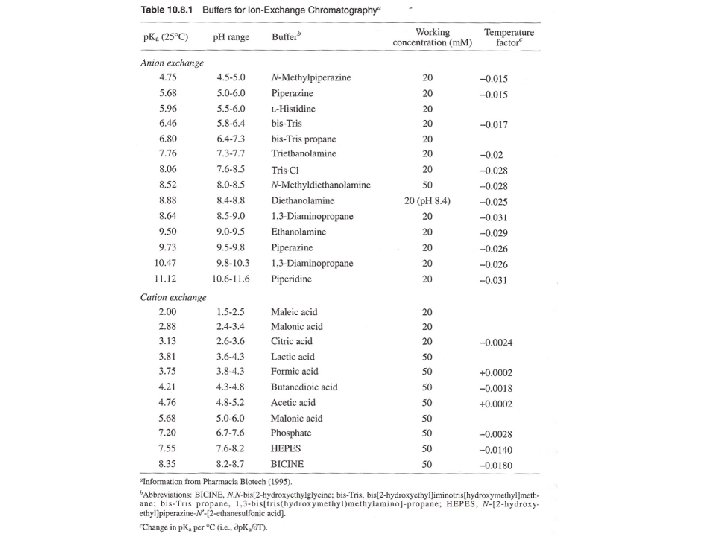
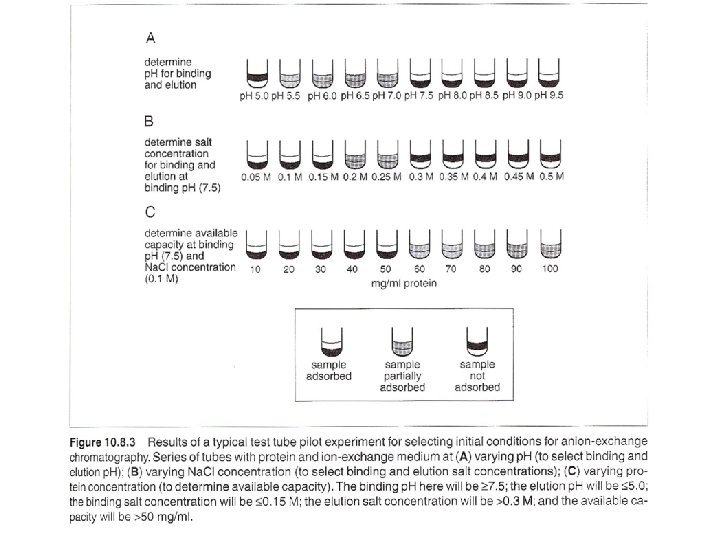
Ion-Exchange Chromatography A. Selecting a buffer system If the p. I of the target protein is known, use anion-exchange medium with operating p. H > p. I, or cation-exchange medium with operating p. H < p. I. If the p. I is unknown, determine the p. I by isoelectric focusing (1 -D of 2 -D electrophoresis). The optimal p. H for binding, elution, and the binding capability at the optimal condition can be determined. Most proteins in cells has p. I below p. H 7, can start an anion- exchange medium and operating p. H of 8. 5 to start, then evaluate the results and optimize conditions. .
B. Selecting a buffer system - Consider the p. H stability of the sample. - Anionic buffers for cation exchange, - Cationic buffers for anion exchange. (Thus, buffering ions will not bind gel matrix)
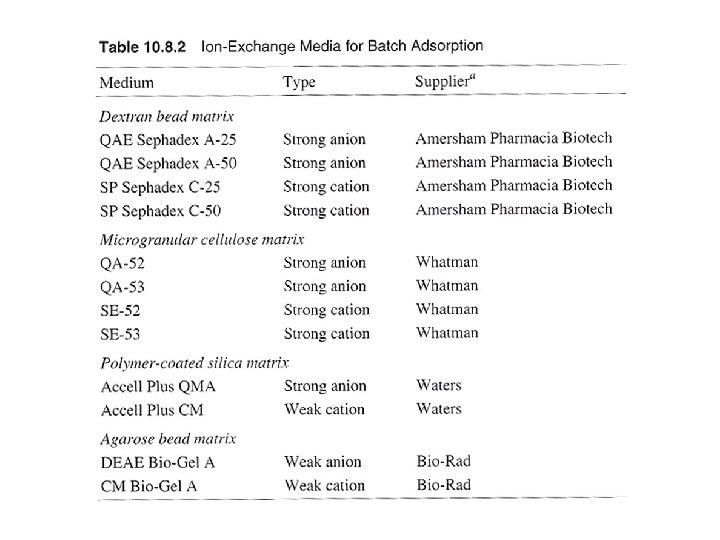
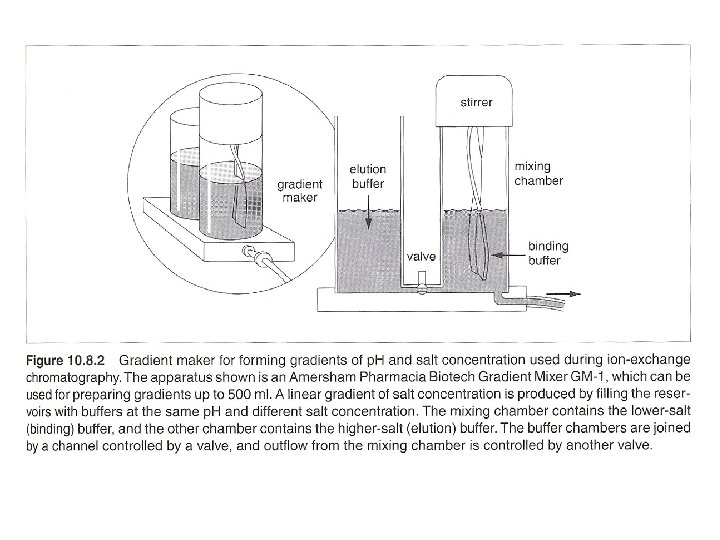
Batch adsorption and step-gradient elution (with increasing salt concentration) eg. anion exchange gel, binding buffer: 20 m. M Tris. Cl, p. H 7. 5 washing buffer: 20 m. M Tris. Cl, p. H 7. 5/100 m. M Na. Cl elution buffer: 20 m. M Tris. Cl, p. H 7. 5/350 m. M Na. Cl regeneration buffer: 20 m. M Tris. Cl, p. H 7. 5/ 2 M Na. Cl
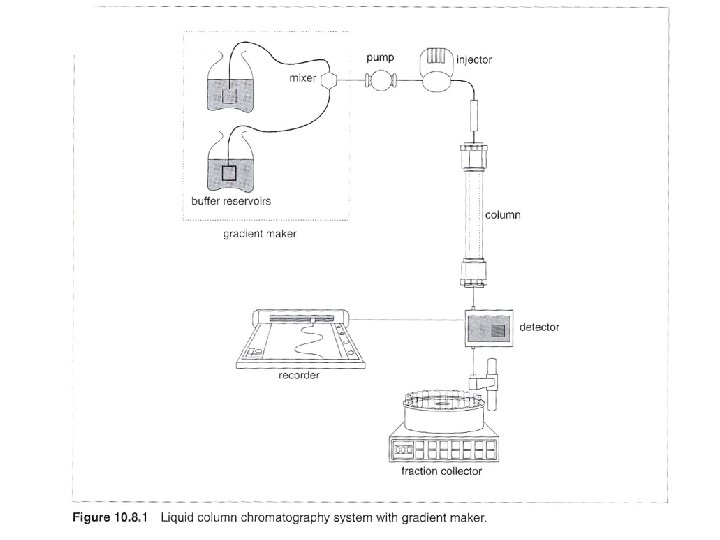
Column chromatography with linear gradient elution eg. 1 ml bed-volumn anion exchange column, binding buffer: 20 m. M Tris. Cl, p. H 7. 5 elution buffer: 20 m. M Tris. Cl, p. H 7. 5/ 1 M Na. Cl 1. Wash column: run 5 Vc volumn of elution buffer at 5 ml/min. 2. Equilibrium: run 5 -10 Vc volumn of binding buffer at 5 ml/min. 3. Collect one fraction at the end, measure p. H and conductivity, see if the same as binding buffer. 4. Filter sample, use total proteins of 25 mg for initial loading. 0. 22 u. M filter for beads < 34 um, 0. 45 um filter for beads between 34 um and 90 um, 1 um filter for beads > 90 um
5. Open injection valve for sample injection, and begin to collect fractions at 1 ml. Can reduce flow rate for very concentrated sample, or increase flow rate for very diluted sample. 6. After sample injected, wash with 3 to 5 Vc volumn of binding buffer at flow rate 5 ml/min. Monitor signal to baseline. 7. Close sample injection valves to reduce system dead volumn. 8. Elute with a linear gradient from 0% to 100% elution buffer in 20 Vc (20 ml). 9. Regenerate colunmn by washing with 5 Vc of elution buffer. 10. Reequilibrate column with 5 to 10 Vc of binding buffer.
Preparation of antibody-sepharose Covalently linking an antibody to Sepharose (CL-4 B, or CL-2 b for high MW antigen), using CNBr activation method. 1. dialyze 1 -30 m g/ml antibody against 0. 1 M Na. HCO 3 /0. 5 M Na. Cl at 40 C with 3 buffer changes over 24 hrs, use dialysis solutions 500 times of the antibody volumn. 2. Centrifuge 3. Measure A 280 (mg/ml Ig. G =A 280 / 1. 44), dilute to 5 mg/ml with 0. 1 M Na. HCO 3 /0. 5 M Na. Cl 4. Wash the sepharose with 10 vol water, use Waterman no. 1 filter and Buchner funnel. 5. Add equal volumn of 0. 2 M Na 2 CO 3 to Sepharose. 6. Add CNBr/ acetonitrile dropwise (in hood). 7. Filter, dry, and add 0. 1 M HCl, and add antibody solution, for 2 hr at rt. 8. Add glycine to saturate the active group on Sepharose, and measure A 280 to determine percentage coupling