Alexei Fedorov Ph D Associate Professor Head of
















 The bottom")





 • Frequency less than 0. 2% across all")






 Nx = 303,")





- Slides: 37
Alexei Fedorov, Ph. D. Associate Professor Head of Bioinformatics Lab Department of Medicine Vice Director Program in Bioinformatics and Genomics/Proteomics Tel: (419)‑ 383‑ 5270 Email: alexei. fedorov@utoledo. edu http: //bpg. utoledo. edu/~afedorov/lab/ 1
Accomplishment of “ 1000 Genome Project” revealed immense amount of information about variation, mutation dynamics, and evolution of the human DNA sequences. • Genomes of thousands patients have been sequenced • 1092 genomes are publicly available (~10 x coverage) • Genomes of 12 famous humans are available(Jim Watson; Craig Venter) (~50 x coverage)
THANKS Ahmed Al-Khudhair, Shuhao Qiu, Meghan Wyse, Shilpi Chowdhury, Xi Cheng, Dulat Bekbolsynov, Arnab Saha-Mandal, Larisa Fedorova
Wikipedia
Presently: Hi. Seq X Ten
In the nearest future
“ 1000 Genomes” international project
THE 1000 GENOME PROJECT q ASW African Americans q YRI Nigeria q LWK Kenya q q q CEU TSI FIN GBR IBS Europeans in America Italy Finland Great Britain Spain q CHB Chinese in Beijing q CHS Chinese from South q JPT Japan q CLM Colombians q MXL Mexicans q PUR Puerto Rican CHB LWK FIN CHS JPT YRI GBR 1000 Genome Project ASW TSI CEU PUR CLM IBS MXL
How genome sequence looks like? 38. 2 M SNPs; 3. 9 M short insertions/deletions; and 14 K deletions
Human chromosome 1 4, 814, 628 lines = =100, 000 pages = 100 books (1000 pages each) 12
Number Of Pairs of Individuals distribution of the number of genetic variants among pairs of individuals from the same 1000 5 European population 900 3 Asian Populations 800 Populations 3 African Population s 700 600 500 400 3 American Populations 200 100 0 2. 7 2. 9 3. 1 3. 3 3. 5 3. 7 3. 9 4. 1 4. 3 4. 5 4. 7 4. 9 5. 1 5. 3 5. 5 Number of Differences * 1, 000 BOTTOM LINE: Two individuals, even from the same population, differ from one another by millions of SNPs
Differences between populations from different continents 1400 LWK 1000 FIN 800 CHB 600 400 LWK vs CHB 200 LWK vs FIN Number of total genomic variants differences (in millions) 5. 7 5. 6 5. 5 5. 4 5. 3 5. 2 5. 1 5 4. 9 4. 8 4. 7 4. 6 4. 5 4. 4 4. 3 4. 1 4. 2 4 3. 9 3. 8 3. 7 3. 6 3. 5 3. 4 3. 3 3. 2 3. 1 3 2. 9 2. 8 0 2. 7 # of pairs of individuals 1200
i n d i v i d u a l s 1600 CHB VS JPT 1400 CHB 1200 JPT 1000 TSI vs FIN 800 FIN 600 TSI 400 LWK vs YRI 200 YRI LWK Number of total genomic variants differences (in millions) 5. 7 5. 6 5. 5 5. 4 5. 3 5. 2 5. 1 5 4. 9 4. 8 4. 7 4. 6 4. 5 4. 4 4. 3 4. 2 4. 1 4 3. 9 3. 8 3. 7 3. 6 3. 5 3. 4 3. 3 3. 1 3. 2 3 2. 9 2. 8 0 2. 7 o f s Differences between populations from the same continent
Why the peaks are narrow?
Major haplotypes of human hemeoxygenase-1 gene (only of frequent SNPs are shown) The bottom line: Mutations never exist alone but in groups linked with each other and forming haplotypes that slowly change due to meiotic recombination and selection/drift
4 th SNP of red haplotype in AGT responsible for myocardial infarction. AGT gene
Linkage disequilibrium
Why the peaks are narrow? Number of loci With equilibrium
Wikipedia: Coefficient of relationship http: //en. wikipedia. org/wiki/Co efficient_of_relationship
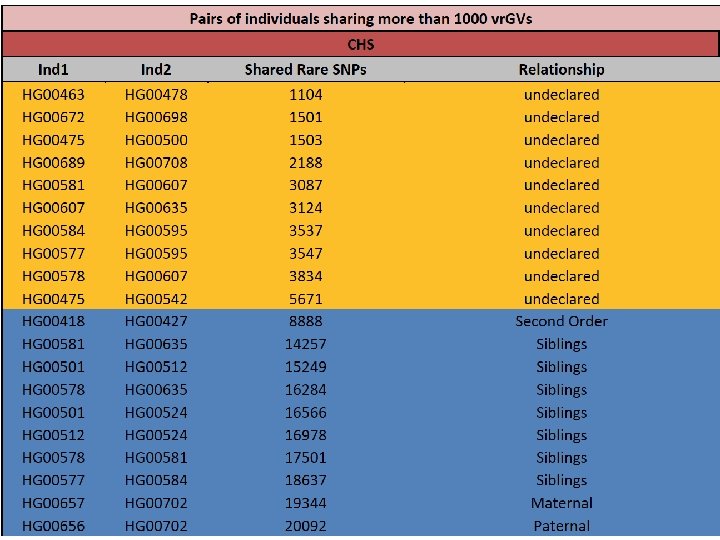
Finding distant genetic relationships • A majority of genomic differences between pairs of individuals is contributed by frequent SNPs that form several (usually from two to five) major haplotypes in each loci. These major haplotypes have a high probability of being the same between genetically non-related individuals. • This obstacle can be overcome if we consider only the very rare SNPs, for which probabilities of being shared by non-related individuals drop dramatically.
Very rare genetic variants (vr. GVs) • Frequency less than 0. 2% across all populations. • Number of vr. GVs per individual in different populations Africa Asia Europe 67, 000 ± 7, 500 24, 100 ± 4, 100 16, 200 ± 2, 700
Number of shared vr. GVs in human pairs from the same population
What is the chance for sharing vr. GVs for non-related individuals? Monte-Carlo computer simulations
Number of shared vr. GVs between populations • The median number of shared vr. GVs was 2 (for CHB-GBR populations), 6 (LWK-FIN), and 8 (for LWK-JPT) • 44, 278 studied pairs formed by individuals from two different continents have less than 118 shared vr. GVs. The highest number of shared vr. GVs between LWK and JPT is 37; LWK-FIN is 80; and GBR-CHB is 117 • The number of shared vr. GVs between populations from the same Asian or European continent is also low (for instance, maximal number between GBR and FIN is 159 and between CHB and JPT is 78)
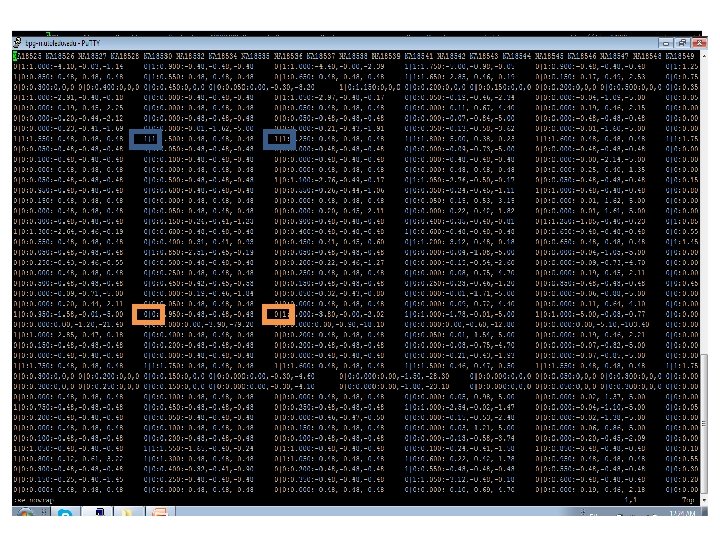
Table 1. Distribution of numbers of shared vr. GVs for 8633 human pairs, where one person of a pair represents British population (GBR), while another person – Chinese population (CHB). *NOTES: detail characterization of shared vr. GVs for the pair, which has 30 shared vr. GVs, is shown in the Table 2. Detail characterization of shared vr. GVs for three pairs at the bottom of this table (marked by *) is shown in the Supplementary Table S 5.
Table 2. Characterization of 30 shared vr. GVs for the British-Chinese pair composed by HG 00255 and NA 18614 individuals. Those vr. GVs that are located in the same locus on chromosome 11 are shaded.
How to calculate the coefficient of relationship from the number of shared vr. GVs (Nx)? Use reference point – the number of shared vr. GVs for siblings (N 50) X%/(Nx –Npeak) = 50%/(N 50 –Npeak) • Npeak is the number of shared vr. GVs corresponding to the peak value (Npeak approximates an average number of shared vr. GVs for genetically non-related pairs) • Nx is the number of shared vr. GVs for the pair under examination ONLY FOR CLOSE RELATIVES !
Example of calculation of coefficient of relationship for Chinese population (CHS) Nx = 303, Npeak =100 R = 0. 59% Third cousins R=0. 78%
Number of shared vr. GVs between African populations NA 19443– NA 18508 pair for LWK-YRI has 1121 shared vr. GVs (R =1. 3%) NA 19350 - NA 20348 and NA 19397 - NA 20348 pairs for LWK-ASW have 903 and 939 of shared vr. GVs respectively (R= 1. 0%)
Nucleotide sequence differences on the whole-genome scale have been computed for 1092 people from 14 populations publicly available by the 1000 Genomes Project. Total number of differences in genetic variants between 96, 464 human pairs has been calculated. The distributions of these differences for individuals within European, Asian or African origin were characterized by narrow unimodal peaks with mean values of 3. 8, 3. 5, and 5. 1 million respectively and standard deviations of 0. 1 -0. 03 million. The total numbers of genomic differences between pairs of all known relatives were found to be significantly lower than their respective population means and in reverse proportion to the distance of their consanguinity. By counting the total number of genomic differences it is possible to infer familial relations for people that share down to 6% of common loci identicalby-descent. Detection of familial relations can be radically improved when only very rare genetic variants (with frequencies less than 0. 2%) are taken into account. Counting of total number of shared very rare SNPs from whole-genome sequences allows establishing distant familial relations for persons with 8 th and 9 th degree of relationship. Using this analysis we predicted 271 distant familial pair-wise relations among 1092 individuals that have not been declared by 1000 Genomes Project. With affordable whole-genome sequencing techniques, very rare SNPs should become important genetic markers for familial relationships and population stratification.
Dynamics of vr. GVs • On average, each person has from 40 to 100 novel mutations that are absent in the genome his/her parents. • An intense influx of novel mutations is an important endless source for vr. GVs, which pool continuously renovates and maintains at a very high level (14 -40 thousand vr. GVs per individual in European and Asian populations).
CONCLUSIONS Application of vr. GVs analysis for obtaining distant genetic relations could be a valuable molecular genetic technique in criminal investigations, in civil familial searching as well as for population, clinical and association studies.
Homework 1. Read the paper: Al-Khudhair A, Qiu S, Wyse M, Chowdhury S, Cheng X, Bekbolsynov D, Saha-Mandal A, Dutta R, Fedorova L, Fedorov A. Inference Of Distant Genetic Relations In Humans Using "1000 Genomes". Genome Biol Evol. 2015 Jan 7. pii: evv 003. [Epubahead of print] Pub. Med PMID: 25573959. 2. Read to reviews on this paper from the first submission and the revision. Write an answer to these reviews (about one page long).