Diagnostyka zaburze krzepnicia krwi Katarzyna KapelkoSowik Stany zakrzepowozatorowe

Diagnostyka zaburzeń krzepnięcia krwi Katarzyna Kapelko-Słowik
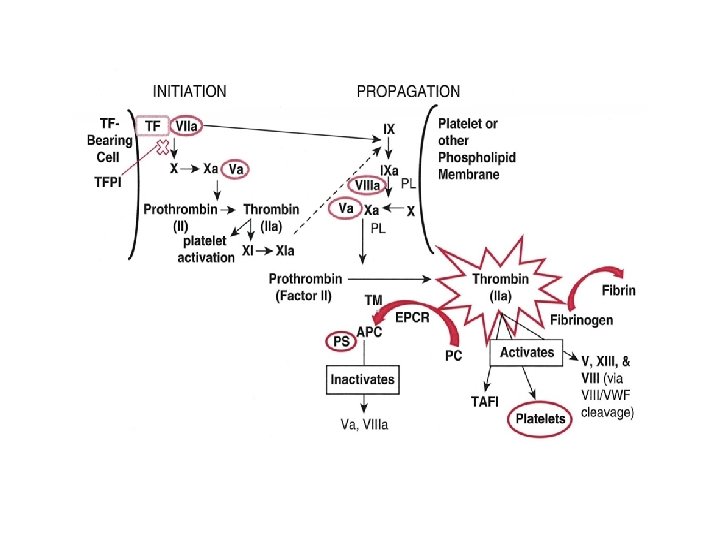

Stany zakrzepowo-zatorowe Zakrzepica jest wynikiem zachwiania równowagi hemostatycznej pomiędzy naturalnymi układami antykoagulacyjnymi (ukł. endogennych inhibitorów, degradacja cz. krzepnięcia i sprawna fibrynoliza) a czynnikami sprzyjającymi aktywacji krzepnięcia. Wrodzona trombofilia Nabyta trombofilia Bezruch Stan zapalny Estrogeny Zabiegi chirurgiczne Nowotwór złośliwy zakrzepica
 -białko C -białko S • Endogenne")
Stany zakrzepowo-zatorowe • Naturalny układ antykoagulacyjny: -trombomodulina (TM) -białko C -białko S • Endogenne inhibitory krzepnięcia: -Antytrombina (AT) TFPI (inhibitor TF ) blokuje kompleks cz. VIIa-TF Kofaktor heparyny II (HCII) -alfa 1 -antytrypsyna -PGI 2 -EDRF (śródbłonkowy czynnik wazodilatacyjny, NO) -INH C 1 - inhibitor kalikreiny osoczowej
 Wrodzony niedobór AT")
Stany zakrzepowo-zatorowe Najczęstsze przyczyny wrodzonej trombofilii Zjawisko oporności na APC (APC-R) Wrodzony niedobór AT Niedobór białka C Niedobór białka S Polimorfizm G 20210 A genu protrombiny Polimorfizm genu MTHFR (hiperhomocysteinemia) Niedobór HC II (heparin kofactor II) Niedobór czynnika Hagemana, cz. XII (APTT ) Zwiększona aktywność czynnika VIII, IX, XI Dysfibrynogenemia Polimorfizm genu dla PAI-1 -
 charakteryzuje się występowaniem przeciwciał antyfosfolipidowych (a. PL)")
Stany zakrzepowo-zatorowe Nabyta trombofilia: Zespół antyfosfolipidowy (APS) charakteryzuje się występowaniem przeciwciał antyfosfolipidowych (a. PL) lub antykardiolipinowych (a. CL) w surowicy z obecnością objawów klinicznych Klasyfikacja APS -pierwotny (46%) -wtórny a- w przebiegu SLE-18% b- w przebiegu innych chorób autoimmunologicznych (typ lupus like)-19% c- towarzyszy ch. nowotworowym- 17% Kryteria diagnostyczne APS: • Kryteria laboratoryjne: LA, p/ciała antykardiolipinowe- Ig. G, Ig. M, p/ciała przeciw beta 2 –glikoproteinie Ig. G, Ig. M • Kryteria kliniczne; zakrzepica żylna/tętnicza Niepowodzenia położnicze (1 x>10 tyg lub 3 x <10 tyg ciąży)

Stany zakrzepowo-zatorowe DIAGNOSTYKA ZAKRZEPICY: 1. badanie kliniczne: -wywiad rodzinny -zakrzepica w młodym wieku -nawracające stany zakrzepowe -wędrujące zakrzepowe zapalenie żył powierzchownych Badania dodatkowe: -angiograficzne (kontrastowe i izotopowe) -przepływowe (met. Dopplera) -pletyzmograficzne TESTY PRZESIEWOWE -morfologia krwi-PT (INR 0. 94 -1. 1)-APTT (37 -46 s)-TT (15 s)-stężenie fibrynogenu (2 -4 g/l), aktywność AT, aktywność białka S, białka C i APR-C
 3. Cytoredukcyjne")
Stany zakrzepowo-zatorowe • LECZENIE: 1. trombolityczne 2. p/zakrzepowe (heparyna, LWMH, VKA, DOAC) 3. Cytoredukcyjne (cytostatyki w nadpłytkowości) 4. Antyagregacyjne (ASA, klopidogrel)
 Zagrożenie zakrzepowe")
Pierwotna i wtórna profilaktyka przeciwzakrzepowa u osób obarczonych trombofilią (rekomendacja ACCP 2012/2016) Zagrożenie zakrzepowe Postępowanie Duże ryzyko n dwa lub więcej incydentów samoistnej zakrzepicy n jeden incydent zakrzepicy zagrażającej życiu (masywny zator tętnicy płucnej, zakrzepica żył mózgowych, krezkowych lub żyły wrotnej ) n jeden incydent samoistnej zakrzepicy u osoby z przeciwciałami antyfosfolipidowymi, niedoborem antytrombiny, więcej niż jednym defektem genetycznym i u homozygot czynnika V Leiden, gen G 20210 A protrombiny n inne trombofilie przy b. obciążonym wywiadzie rodzinnym Umiarkowane ryzyko n jeden incydent zakrzepicy wtórnej (np. po urazie, zabiegu operacyjnym, podczas ciąży) n bezobjawowi nosiciele defektu Bezterminowa antykoagulacja Profilaktyka wtórna przez 3 -6 miesięcy, profilaktyka pierwotna w sytuacjach grożących zakrzepicą n Profilaktyka pierwotna w sytuacjach grożących zakrzepicą. n

Skazy krwotoczne 1. 2. 3. 4. Skazy krwotoczne naczyniowe Skazy krwotoczne płytkowe Skazy krwotoczne osoczowe min. hemofilia A, B, choroba von Zaburzenia fibrynolizy

Skazy krwotoczne-diagnostyka
 N - PT, APTT PT, N")
Wstępna diagnostyka pacjenta ze skazą krwotoczną (test korekcji) N - PT, APTT PT, N - APTT PT, APTT Zmieszać 50: 50 osocze badane i osocza prawidłowe N N N deficyt VIII, IX, XII 50: 50 Inhibitor: VIII, IX, XI APS - często deficyt VII rzadko 50: 50 Inhibitor: VII (rzadko) APS - rzadko deficyt X, V, II, I rzadko 50: 50 Inhibitor: X, V, II, I (rzadko) APS - często N - PT, N - APTT Dysfibrynogenemia Niedobór cz. XIII Deficyt a 2 -plazminy Niewielki niedobór cz. krzepnięcia >25%<40% – FDP Gammapatia monoklonalna Płytki krwi – zmniejszona liczba lub zmieniona jakość, Zaburzenia naczyniowe

Hemofilia A i B Ø zaburzenie krzepnięcia krwi - wynik wrodzonego niedoboru czynników VIII i IX, Ø po raz pierwszy wspominano o tej chorobie już w II wieku naszej ery w Talmudzie, Ø dokładniej znana od 1800 r. Ø w 1872 r - hemofilię to trwająca całe życie skłonność do wylewów dostawowych i domięśniowych, Ø na początku XX wieku - diagnozowana wyłącznie na podstawie charakterystycznych krwawień i historii rodzinnej, Ø 1940 -1950 wyizolowano z osocza cz. VIII i cz. IX, Ø niedobór cz. IX nazwano chorobą Christmasa (od nazwiska kanadyjskiego chłopca, u którego po raz pierwszy wykryto to zaburzenie), Ø w 1970 r odróżniono hemofilię A od choroby von Willebranda.

Diagnostyka hemofilii • • • Rozpoznanie hemofilii ważny jest wywiad rodzinny ale u 20 -30% noworodków urodzonych z hemofilią nie stwierdzało się w rodzinie tej choroby UWAGA! Osoby z łagodną postacią choroby mogą nie manifestować objawów hemofilii po urodzeniu Jeśli aktywność cz. IX jest < 1% normy, rozpoznanie ciężkiej hemofilii jest pewne. U zdrowego noworodka aktywność cz. VIII jest porównywalna z aktywnością cz. VIII u zdrowego dorosłego. U chorych na ciężką i umiarkowaną hemofilię aktywność cz. VIII i cz. IX pozostaje na tym samym poziomie przez całe życie ( wyjątek hemofilia B Leyden). W łagodnej hemofilii A stwierdza się krótkotrwałe zwiększenie aktywności

Hemofilia A/B Hemofilia A 1 na 5000 męskich urodzeń gen cz. VIII ma 186 kb zawiera 0, 001 DNA chromosomu X i jest jednym z największych zidentyfikowanych ludzkich genów znaleziono > 150 różnych mutacji, 70 % rodzin ma różne mutacje, Øw 90 % przyczyna mutacji może być wykryta dla umiarkowanej i łagodnej hemofilii, a tylko w 50 -60% dla ciężkiej hemofilii Hemofilia B częstość ¼-1/6 zachorowań na hemofilię A, gen dla cz. IX ma 34 kb zlokalizowany centromerycznie do genu cz. VIIIw końcowym długim ramieniu chromosomu X, nie znaleziono połączenia między genami dlacz. VIII i IX, nosicielstwo hemofilii B wykrywa się w 60 -70 % mierząc obniżenie aktywności cz. IX w surowicy, skuteczniejsza w wykrywaniu nosicielstwa jest analiza mutacji genu.

Zasady leczenia substytucyjnego • Wyróżnia się 3 rodzaje rekombinowanych koncentratów cz. VIII/ cz. IX: 1. Pierwszej generacji, w których stabilizatorem jest albumina, białka zwierzęce są dodawane do podłoża hodowlanego 2. Drugiej generacji, w których stabilizatorem są związki cukru i białka zwierzęce są obecne 3. Trzeciej generacji – pozbawione białek ludzkich i zwierzęcych Leczenie: Ø zależy od typu i rozwoju krwawienia i wynosi średnio 30 -50 % normalnego poziomu, co wystarcza dla zabezpieczenia większości epizodów krwawienia, ale Ø 50 -100% normalnego poziomu potrzeba do leczenia i zapobiegania krwawieniom zagrażającym życiu, 1. jak uzupełniać? 2. np. cz. VIII jako bolus co 8 -12 h lub ciągła infuzja utrzymująca stały terapeutyczny poziom czynnika

Rodzaje profilaktyki stosowanej w hemofilii A/B • • • Profilaktyka pierwotna: Podawanie cz. VIII lub cz. IX rozpoczęte przed lub po wystąpieniu pierwszego krwawienia do stawu i przed ukończeniem 2 rż, w celu prewencji artropatii; czas stosowania – do ukończenia wzrostu kostnego. Profilaktyka wtórna: Podawanie cz. rozpoczęte po wystąpieniu >2 krwawień do stawu lub w wieku >2 lat, w celu zmniejszenia tempa postępu artropatii – do ukończenia 18 rż Profilaktyka krótkoterminowa: Regularne podawanie cz. w celu zahamowania powtarzających się krwawień do stawu – stosowanie przez kilka tygodni- miesięcy • Powikłania terapii substytucyjnej: • Hepatitis C w 70 - 80% przebiega bezobjawowo; dopiero biopsja wątroby wykazuje w 70 % cechy przewlekłego zapalenia wątroby, a w 15 % marskość wątroby. HIV Choroba Creutzfeldta- Jacoba
- Slides: 17