Molecular Docking G Schaftenaar Docking Challenge Identification of


 space • Scoring")

 Molecular Dynamics (MD)")
 • Make random move to")










")

 é Aij Bij qi q j ù ú = åå")


 ij æ i ö ( ) s seg r")



- Slides: 26
Molecular Docking G. Schaftenaar
Docking Challenge • Identification of the ligand’s correct binding geometry in the binding site (Binding Mode) • Observation: – Similar ligands can bind at quite different orientations in the active site.
Two main tasks of Docking Tools • Sampling of conformational (Ligand) space • Scoring protein-ligand complexes
Rigid-body docking algorithms • Historically the first approaches. • Protein and ligand fixed. • Search for the relative orientation of the two molecules with lowest energy. • FLOG (Flexible Ligands Oriented on Grid): each ligand represented by up to 25 low energy conformations.
Introducing flexibility: Whole molecule docking • • Monte Carlo methods (MC) Molecular Dynamics (MD) Simulated Annealing (SA) Genetic Algorithms (GA) Available in packages: Auto. Dock (MC, GA, SA) GOLD (GA) Sybyl (MD)
Monte Carlo • Start with configuration A (energy EA) • Make random move to configuration B (energy EB) • Accept move when: EB < EA or if EB > EA except with probability P:
Molecular Dynamics • force-field is used to calculate forces on each atom of the simulated system • following Newton mechanics, calculate accelerations, velocities and new coordinates from the forces. (Force = mass times acceleration) • The atoms are moved slightly with respect to a given time step
Simulated Annealing Finding a global minimium by lowering the temperature during the Monte Carlo/MD simulation
Genetic Algorithms • Ligand translation, rotation and configuration variables constitute the genes • Crossovers mixes ligand variables from parent configurations • Mutations randomly change variables • Natural selection of current generation based on fitness • Energy scoring function determines fitness
Introducing flexibility: Fragment Based Methods • build small molecules inside defined binding sites while maximizing favorable contacts. • De Novo methods construct new molecules in the site. • division into two major groups: – Incremental construction (Flex. X, Dock) – Place & join.
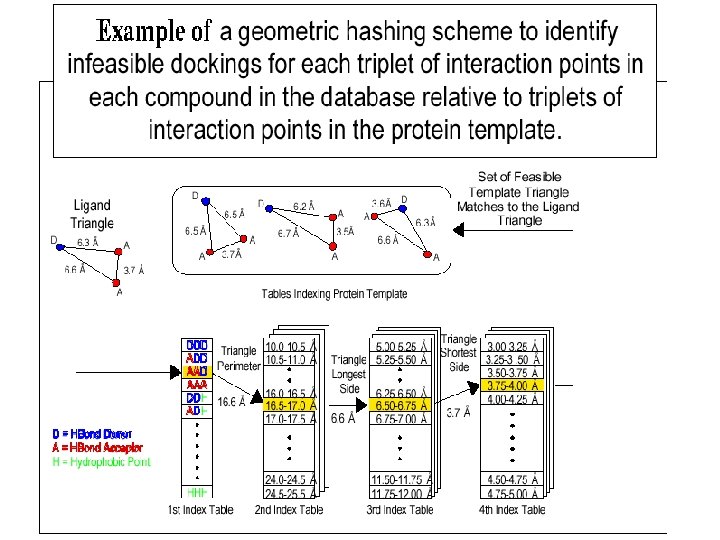
Placing Fragments and Rigid Molecules • All rigid-body docking methods have in common that superposition of point sets is a fundamental sub-problem that has to be solved efficiently: – Geometric hashing – Pose clustering – Clique detection
Geometric hashing • originates from computer vision • Given a picture of a scene and a set of objects within the picture, both represented by points in 2 d space, the goal is to recognize some of the models in the scene
Pose-Clustering • For each triangle of receptor compute the transformation to each ligand matching triangle. • Cluster transformations. • Score the results.
Clique-Detection • • Nodes comprise of matches between protein and ligand • Edges connect distance compatible pairs of nodes • In a clique all pair of nodes are connected
Scoring Functions • Shape & Chemical Complementary Scores • Empirical Scoring • Force Field Scoring • Knowledge-based Scoring • Consensus Scoring
Shape & Chemical Complementary Scores • Divide accessible protein surface into zones: – Hydrophobic – Hydrogen-bond donating – Hydrogen-bond accepting • Do the same for the ligand surface • Find ligand orientation with best complementarity score
Empirical Scoring parameters fit to reproduce Measured binding affinities (Flex. X, LUDI, Hammerhead)
Empirical scoring DG = DG 0 + DGrot ´ N rot + DGhb + DGio å f (DR, Da ) Loss of entropy during binding Hydrogen-bonding neutral. H -bonds Ionic interactions ionic -int. + DGarom Aromatic interactions arom. int + DGlipo å f (DR, Da ) lipo. cont. Hydrophobic interactions
Force Field Scoring (Dock) é Aij Bij qi q j ù ú = åå ê 12 - 6 + c rij r ij úû i j ê ë rij lig prot Enonbond Nonbonding interactions (ligand-protein): -van der Waals -electrostatics Amber force field
Knowledge-based Scoring Function Free energies of molecular interactions derived from structural information on Protein-ligand complexes contained in PDB Boltzmann-Like Statistics of Interatomic Contacts. [ ] P (s p , s l )= Pref exp - b. F (s p , s l )
Distribution of interatomic distances is converted into energy functions by inverting Boltzmann’s law. F P(N, O)
Potential of Mean Force (PMF) ij æ i ö ( ) s seg r Fij (r ) = - k BT lnçç f. Vol _ corr (r ) ij ÷÷ s bulk ø è s ij seg (r ) ij s bulk Number density of atom pairs of type ij at atom pair distance r Number density of atom pairs of type ij in reference sphere with radius R
Consensus Scoring Cscore: Integrate multiple scoring functions to produce a consensus score that is more accurate than any single function for predicting binding affinity.
Virtual screening by Docking • Find weak binders in pool of nonbinders • Many false positives (96 -100%) • Consensus Scoring reduces rate of false positives
Concluding remarks Scoring functions are the Achilles’ heel of docking programs. False positives rates can be reduced using several scoring functions in a consensus-scoring strategy Although the reliability of docking methods is not so high, they can provide new suggestions for protein-ligand interactions that otherwise may be overlooked