GerstmannStrusslerScheinker disease Fabrizio Tagliavini Fondazione IRCCS Istituto Neurologico

Gerstmann-Sträussler-Scheinker disease Fabrizio Tagliavini Fondazione IRCCS – Istituto Neurologico “Carlo Besta” Milano
 • Creutzfeldt-Jakob disease - sporadic")
Human Prion Diseases • Kuru - acquired (ritualistic cannibalism) • Creutzfeldt-Jakob disease - sporadic - genetic - acquired (iatrogenic, new variant) • Gerstmann-Sträussler-Scheinker Disease - genetic • Fatal Insomnia - genetic - sporadic

Z. Neurol. 154: 736 -762, 1936 Berta H • Onset • Clinical • Death • Family • 26 yrs, gait disturbances picture: severe ataxia, dysmetria, dysartria, Babinski sign, personality and behavioural changes 31 yrs history: several family members showed a similar disease, with onset in the 3 rd-4 th decade and duration of 6 -8 yrs, consistent with autosomal dominant transmission Clinical diagnosis: Hereditary Spino-Cerbellar Ataxia

• Cerebellum: loss of granule cells and severe degeneration of dentate nuclei • Spinal Cord: pallor of spinocerebellar tracts and posterior columns • Argentophilic plaques in the cerebellar cortex, cerebral cortex and basal ganglia • Vacuolar changes in layers IIIII of cerebral cortex Diagnosis: peculiar form of degenerative spino-cerebellar atrophy

Gerstmmann-Sträussler-Scheinker disease • Successfull transmission to primates: spongiform encephalopathy (Masters et al. , Brain 1981) Prion Disease • Amyloid plaques are composed of Prion Protein • P 102 L mutation in the PRNP gene in affected members of the original family (Hainfellner et al. , Brain Pathol. 1995) • Phenotypic variability even within the same familiy (ataxia vs dementia); cases with rapid course and more pronounced spongiform changes (CJD-like picture)

Phenotypic heterogeneity in GSS P 102 L Typical form CJD-like form T 1 DWI

Genetics Systematic analysis of the PRNP gene in neurodegenerative disorders has enabled to recognize several new GSS variants with different clinical phenotype

PRNP mutations associated with GSS PRNP mutation Main Clinical Feature Age and Duration No. Fam. > 30 P 102 L Cerebellar Syndrome Dementia in late stage III-VI decade, 0. 5 -8 yrs P 105 L Spastic Paraparesis and Dementia IV-V decade, 6 -12 yrs 5 A 117 V Atypical Parkinsonism and Dementia Cerebellar syndrome (1 family) II-VII decade, 1 -11 yrs 8 G 131 V Dementia and Extrapyramidal Signs Ataxia in late stage V decade, 9 yrs 1 H 187 R Dementia and Cerebellar Syndrome IV-VI decade, 7 -18 yrs 1 F 198 S Cerebellar Syndrome, Parkinsonism and Dementia IV-VIII decade, 2 -12 yrs 3 D 202 N Dementia and Cerebellar Syndrome VIII decade, 6 yrs 2 Q 212 P Cerebellar Syndrome VI decade, 8 yrs 1 Q 217 R Dementia and Parkinsonism V-VII decade, 2 -6 yrs 2 M 232 T Cerebellar Syndrome, Spastic Paraparesis and Dementia V decade, 6 yrs 1

Possible basis of phenotypic heterogeneity Distinct Pr. Pres isoforms are associated with different GSS phenotypes s. CJD Type 1 s. CJD Type 2 GSS GSS GSS P 102 L A 117 V G 131 V F 198 S D 202 N Q 217 R Q 212 P 33 33 28 28 21 21 18 18 14 14 8 8

Diagnostic Problems Clinical features: broad spectrum of clinical phenotypes that mimic other neurodegenerative diseases such as spinocerebellar ataxia, parkinsonian syndromes, spastiac paraparesis, or atypical dementia (AD- or FTD-like) In most patients EEG : MRI CSF: CSF no pseudoperiodic bi-triphasic complexes regional atrophy without signal abnormalities • 14 -3 -3 absent or weakly positive • Tau normal or slightly increased Diagnosis is dependent on genetics: PRNP mutation Recent diagnostic tool: PET imaging with amyloid-binding probes (e. g. FDDNP, PIB)

Kepe et al. Brain Pathology 2009

Nosology Ø Only GSS P 102 L has been successfully transmitted to experimental animals (primates and rodents) Ø Several attempts to transmit the other GSS genotypes have been unsuccessful to date Most GSS variants seem to be Pr. P-related neurodegenerative disorders rather than prion diseases
")
Some GSS variants have a neuropathological profile closely similar to Alzheimer’s disease (neurofibrillary tangles) Ø Research on GSS can help understanding the molecular basis of nerve cell degeneration in AD and vice versa Ø Treatments strategies for AD targeting neurofibrillary pathology may be effective in these GSS variants

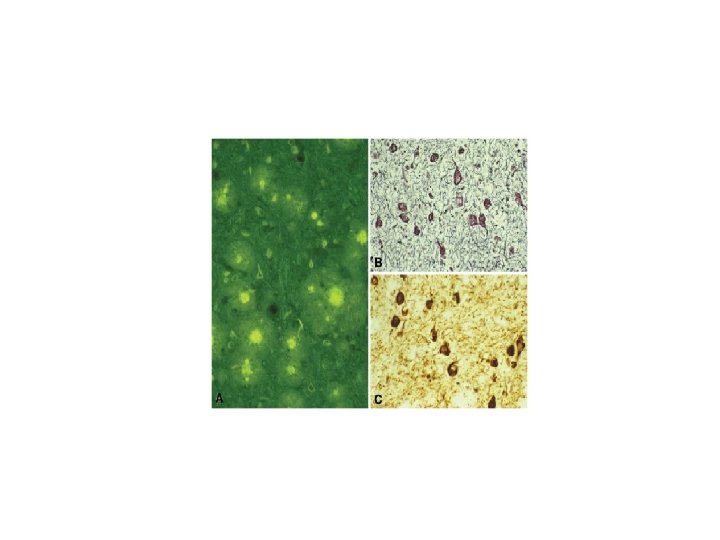
GSS F 198 S-V 129 Onset: IV-VIII decade Onset is 10 years earlier in patients with VV at codon 129 than in patients with MV Clinical Picture: Cerebellar syndrome, parkinsonism, dementia Duration: 2 -12 years Pathology: Pr. P amyloid and neurofibrillary tangles similar to those of Alzheimer’s disease

GSS F 198 S Pr. P/Tau Ph-Tau Thioflavine

Gerstmmann-Sträussler-Scheinker disease Ø Chronic neurodegenerative disorder primarily involving the motor system Ø Needs and management of patients are partly different from those of CJD patients Educational and intervention programs designed to help long-term caregivers

Thank you for your attention and support !


GSS F 198 S-V 129 Silver Thioflavine S Pr. P

GSS F 198 S-V 129 Pr. P Ph-Tau Thioflavine Pr. P/Tau PK Pr. Pres profile - +
")
GSS, Indiana kindred (F 198 S)
 C 1 q SAP")
Microglia Pr. P (3 F 4) C 1 q SAP
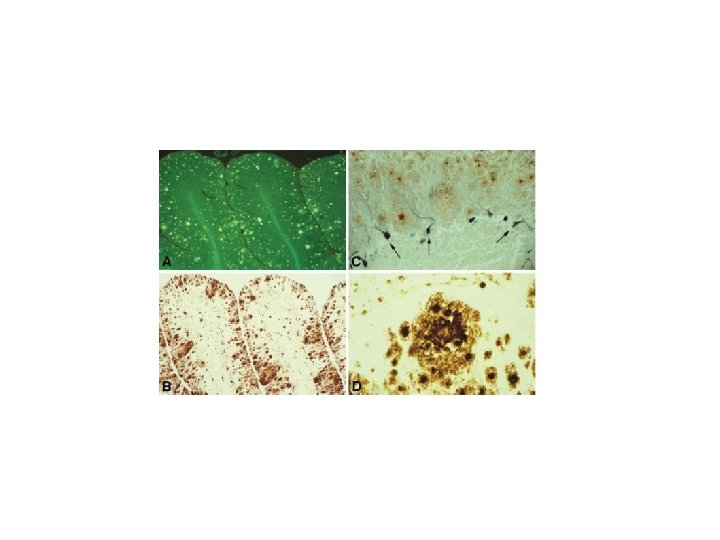

GSS: cerebral amyloidosis Thioflavine S EM Pr. P 23 231 repeats ~ 82 ~146 Amyloid

Pr. PSc profile of GSS differs from that of CJD GSS F 198 S PK PNG - + + + CJD E 200 K - + + +

GSS P 102 L molecular basis of phenotypic variability 1. Codon 129 polymorphism • P 102 L-M 129: Cerebellar syndrome, late Dementia • P 102 L-V 129: Cerebellar syndrome, Seizures, no Dementia, slower course (up to 12 yrs) 2. Pr. PSc type Case 1 PK - + Case 2 - + - 30 k. Da Amyloid + Spongiosis - 8 k. Da
- Slides: 27