Zarzdzanie chemikaliami w przedsibiorstwie ze szczeglnym uwzgldnieniem rozporzdzenia

"Zarządzanie chemikaliami w przedsiębiorstwie ze szczególnym uwzględnieniem rozporządzenia REACH„ Temat: Badania substancji wymagane w ramach REACH i CLP Marek Ubraniak Inspektor DPL/Główny Specjalista Departament do spraw Dobrej Praktyki Laboratoryjnej Biuro do spraw Substancji Chemicznych

REACH • to rozporządzenie Unii Europejskiej przyjęte w celu lepszej ochrony środowiska i zdrowia człowieka przed zagrożeniami, jakie mogą stanowić substancje chemiczne, przy jednoczesnym zwiększeniu konkurencyjności unijnego sektora chemikaliów. Rozporządzenie propaguje również alternatywne metody oceny zagrożeń stwarzanych przez substancje chemiczne w celu ograniczenia liczby badań przeprowadzanych na zwierzętach.

Badania na zwierzętach na mocy REACH • Celem rozporządzenia REACH jest zapewnienie wysokiego poziomu ochrony zdrowia człowieka i środowiska przed wpływem niebezpiecznych chemikaliów. Dąży ono do równowagi: do lepszego zrozumienia możliwych zagrożeń związanych z chemikaliami, a jednocześnie do unikania niepotrzebnych badań na zwierzętach. Aby dowiedzieć się więcej na temat chemikaliów czasami trzeba je w ostateczności przebadać na zwierzętach. Rejestrujący mogą przeprowadzać nowe badania jedynie po wyczerpaniu wszystkich innych stosownych i dostępnych źródeł danych.
")
Definicje i przepis ogólny Artykuł 3 Definicje • W rozumieniu niniejszego rozporządzenia: • 27) pełen raport badawczy: oznacza kompletny i całościowy opis działań wykonanych w celu wygenerowania informacji. Oznacza to kompletną pracę naukową wydaną w formie publikacji z opisem przeprowadzonych badań lub też pełne sprawozdanie sporządzone przez laboratorium i zawierające opis przeprowadzonych badań; • 28) szczegółowe podsumowanie przebiegu badania: oznacza szczegółowe podsumowanie celów, metod, wyników i wniosków pełnego raportu badawczego, dostarczające ilość informacji wystarczającą do przeprowadzenia niezależnej oceny badania i zmniejszające potrzebę korzystania z pełnego raportu badawczego; • 29) podsumowanie przebiegu badania: oznacza podsumowanie celów, metod, wyników i wniosków pełnego raportu badawczego, dostarczające ilość informacji wystarczającą do oszacowania znaczenia badania;

Artykuł 10 Informacje przedkładane dla celów ogólnej rejestracji Dane rejestracyjne wymagane na mocy art. 6 lub art. 7 ust. 1 lub 5 zawierają wszystkie następujące informacje: a) dokumentację techniczną zawierającą: • (v) wytyczne dotyczące bezpiecznego stosowania substancji zgodnie z wymaganiami określonymi w sekcji 5 załącznika VI; • (vi) podsumowania informacji uzyskanych w wyniku zastosowania załączników VII–XI; • (vii) szczegółowe podsumowania informacji uzyskanych w wyniku zastosowania załączników VII–XI, jeżeli jest to wymagane na mocy przepisów załącznika I; • (ix) propozycje przeprowadzenia badań, jeżeli zostały wymienione w załącznikach IX i X;

Artykuł 12 Informacje, które należy przedłożyć w zależności od wielkości obrotu 1. Dokumentacja techniczna, o której mowa w art. 10 lit. a) zawiera w pkt (vi) i (vii) wszelkie dane fizykochemiczne, toksykologiczne i ekotoksykologiczne, które są istotne i dostępne dla rejestrującego, a przynajmniej: • a) informacje określone w załączniku VII w przypadku substancji niewprowadzonych, oraz substancji wprowadzonych, produkowanych lub importowanych przez producenta lub importera w ilości co najmniej 1 tony rocznie i spełniających jedno lub oba kryteria określone w załączniku III; • b) informacje dotyczące właściwości fizykochemicznych określone w sekcji 7 załącznika VII w przypadku substancji wprowadzonych, produkowanych lub importowanych przez producenta lub importera w ilości co najmniej 1 tony rocznie i niespełniających żadnego z kryteriów określonych w załączniku III;
 informacje")
cd. Artykuł 12 Informacje, które należy przedłożyć w zależności od wielkości obrotu c) informacje określone w załącznikach VII i VIII w przypadku substancji produkowanych lub importowanych przez producenta lub importera w ilości co najmniej 10 ton rocznie; d) informacje określone w załącznikach VII i VIII oraz propozycje przeprowadzenia badań w celu dostarczenia informacji określonych w załączniku IX w przypadku substancji produkowanych lub importowanych przez producenta lub importera w ilości co najmniej 100 ton rocznie; e) informacje określone w załącznikach VII i VIII oraz propozycje przeprowadzenia badań w celu dostarczenia informacji określonych w załącznikach IX i X w przypadku substancji produkowanych lub importowanych przez producenta lub importera w ilości co najmniej 1 000 ton rocznie.

cd. Artykuł 12 Informacje, które należy przedłożyć w zależności od wielkości obrotu 2. Kiedy ilość substancji już zarejestrowanej przez producenta lub importera osiągnie kolejny zakres wielkości obrotu, producent lub importer bezzwłocznie informuje Agencję o dodatkowych informacjach, których będzie potrzebował zgodnie z ust. 1. Zastosowanie ma odpowiednio dostosowany art. 26 ust. 3 i 4.

Artykuł 13 Ogólne wymagania dotyczące generowania informacji o swoistych właściwościach substancji 1. Jeżeli wymagania określone w załączniku XI są spełnione, informacje o swoistych właściwościach substancji mogą być generowane w inny sposób niż przez badania. W szczególności w przypadku działania toksycznego dla ludzi informacje są generowane wszędzie tam, gdzie jest to możliwe przy użyciu metod innych niż badania na zwierzętach kręgowych, z wykorzystaniem metod alternatywnych, na przykład metod in vitro lub jakościowych lub ilościowych modeli zależności struktura-aktywność lub na podstawie informacji o substancjach o podobnej strukturze (grupowanie lub podejście przekrojowe). Można pominąć badania wymagane na podstawie sekcji 8. 6 i 8. 7 załącznika VIII, załącznika IX i załącznika X, jeżeli jest to uzasadnione informacjami na temat narażenia i wdrożonymi środkami kontroli ryzyka określonymi w sekcji 3 załącznika XI.

cd. Artykuł 13 Ogólne wymagania dotyczące generowania informacji o swoistych właściwościach substancji • 2. Metody te podlegają regularnym przeglądom i udoskonaleniom w celu zmniejszenia ilości badań przeprowadzanych na zwierzętach kręgowych oraz liczby wykorzystywanych zwierząt. Komisja, po konsultacji z odpowiednimi zainteresowanymi stronami, w razie konieczności przedstawia jak najwcześniej wniosek dotyczący zmiany rozporządzenia Komisji w sprawie metod badawczych przyjętego zgodnie z procedurą, o której mowa w art. 133 ust. 4, a także w razie potrzeby załączników do niniejszego rozporządzenia, tak aby zastąpić, ograniczyć bądź udoskonalić badania na zwierzętach. Poprawki do tego rozporządzenia Komisji przyjmowane są zgodnie z procedurą określoną w ust. 3, a poprawki do załączników do niniejszego rozporządzenia przyjmowane są zgodnie z procedurą określoną w art. 131.

cd. Artykuł 13 Ogólne wymagania dotyczące generowania informacji o swoistych właściwościach substancji • 3. Jeżeli dla wygenerowania informacji o swoistych właściwościach substancji wymagane są badania substancji, przeprowadza się je metodami badań określonymi w rozporządzeniu Komisji lub zgodnie z innymi międzynarodowymi metodami uznanymi za odpowiednie przez Komisję lub Agencję. Komisja przyjmuje to rozporządzenie, którego celem jest zmiana innych niż istotne elementów niniejszego rozporządzenia poprzez uzupełnienie go, zgodnie z procedurą, o której mowa w art. 133 ust. 4. • Informacje o swoistych właściwościach substancji mogą być wygenerowane przy użyciu innych metod badań, pod warunkiem że spełnione są wymagania określone w załączniku XI (OGÓLNE ZASADY DOSTOSOWYWANIA STANDARDOWEGO TRYBU BADAŃ).

cd. Artykuł 13 Ogólne wymagania dotyczące generowania informacji o swoistych właściwościach substancji • 4. Badania i analizy ekotoksykologiczne i toksykologiczne są wykonywane zgodnie z zasadami dobrej praktyki laboratoryjnej przewidzianymi w dyrektywie 2004/10/WE lub innymi międzynarodowymi normami, które Komisja lub Agencja uzna za równoważne, oraz zgodnie z przepisami dyrektywy 86/609/EWG, jeżeli to stosowne.

cd. Artykuł 13 Ogólne wymagania dotyczące generowania informacji o swoistych właściwościach substancji • 5. Jeżeli substancja została zarejestrowana, nowy rejestrujący ma prawo odnieść się do przedłożonych wcześniej podsumowań przebiegu badań lub szczegółowych podsumowań przebiegu badań dotyczących tej samej substancji, pod warunkiem że jest w stanie wykazać, że rejestrowana przez niego substancja jest tą samą substancją, co substancja zarejestrowana wcześniej, z uwzględnieniem stopnia czystości i rodzaju zanieczyszczeń, oraz pod warunkiem że poprzedni rejestrujący wyraził (wyrazili) zgodę na odniesienie się do pełnych raportów badawczych dla celów rejestracji. • Nowy rejestrujący nie odnosi się do tych badań, jeżeli celem jest dostarczenie informacji wymaganych na podstawie sekcji 2 załącznika VI.

TYTUŁ III UDOSTĘPNIANIE DANYCH I UNIKANIE PRZEPROWADZANIA NIEPOTRZEBNYCH BADAŃ ROZDZIAŁ 1 Cele i zasady ogólne Artykuł 25 Cele i zasady ogólne

Artykuł 25 Cele i zasady ogólne 1. W celu unikania badań na zwierzętach badania na zwierzętach kręgowych dla celów niniejszego rozporządzenia przeprowadzane są tylko w ostateczności. Konieczne jest także podejmowanie środków ograniczających powielanie innych badań. 2. Udostępnianie danych i wspólne zgłaszanie informacji zgodnie z niniejszym rozporządzeniem dotyczy danych technicznych, a w szczególności informacji odnoszących się do swoistych właściwości substancji. Rejestrujący nie wymieniają informacji o swej działalności rynkowej, w szczególności informacji dotyczących możliwości wytwórczych, tonażu wytwórstwa lub sprzedaży, tonażu importu lub udziałów w rynku. 3. Wszelkie podsumowania przebiegu badań lub szczegółowe podsumowania przebiegu badań, przedłożone co najmniej 12 lat wcześniej w toku rejestracji dokonywanej na mocy niniejszego rozporządzenia, mogą być wykorzystywane do celów rejestracji przez innego producenta lub importera.

ROZDZIAŁ 2 Zasady dotyczące substancji niewprowadzonych i rejestrujących substancje wprowadzone bez rejestracji wstępnej Artykuł 26 Obowiązek zwrócenia się z zapytaniem przed dokonaniem rejestracji

Artykuł 26 Obowiązek zwrócenia się z zapytaniem przed dokonaniem rejestracji 1. Każdy potencjalny rejestrujący substancję niewprowadzoną lub potencjalny rejestrujący substancję wprowadzoną, który nie dokonał rejestracji wstępnej zgodnie z art. 28, zwracają się do Agencji z zapytaniem o to, czy przedłożono już dokumenty rejestracyjne dla tej samej substancji. Razem z zapytaniem potencjalny rejestrujący przedkłada Agencji następujące informacje: c) informacje o tym, które wymagania w zakresie informacji zobowiązują go do przeprowadzenia nowych badań na zwierzętach kręgowych; d) informacje o tym, które wymagania w zakresie informacji zobowiązują go do przeprowadzenia innych nowych badań. 2. Jeżeli ta sama substancja nie została wcześniej zarejestrowana, Agencja informuje o tym potencjalnego rejestrującego.

cd. Artykuł 26 Obowiązek zwrócenia się z zapytaniem przed dokonaniem rejestracji 3. Jeżeli ta sama substancja została zarejestrowana nie dawniej niż w ciągu ostatnich 12 lat, Agencja informuje niezwłocznie potencjalnego rejestrującego o imionach i nazwiskach lub nazwach oraz adresach poprzednich rejestrujących i o odpowiednich przedłożonych już przez nich podsumowaniach lub szczegółowych podsumowaniach przebiegu badań, zależnie od okoliczności. Badań na zwierzętach kręgowych nie powtarza się.

Artykuł 27 Udostępnianie istniejących danych w przypadku substancji zarejestrowanych 1. W przypadku gdy substancja została zarejestrowana nie dawniej niż w ciągu ostatnich 12 lat, o czym mowa w art. 26 ust. 3, potencjalny rejestrujący: a) zwraca się, w przypadku informacji dotyczących badań na zwierzętach kręgowych; oraz b) może zwrócić się, w przypadku informacji niedotyczących badań na zwierzętach kręgowych, do poprzednich rejestrujących z prośbą o informacje, których potrzebuje w związku z art. 10 lit. a) pkt (vi) i (vii) w celu dokonania rejestracji. 4. Po osiągnięciu porozumienia w zakresie udostępnienia informacji poprzedni rejestrujący udostępnia nowemu rejestrującemu uzgodnioną informację i udziela nowemu rejestrującemu pozwolenia na odwoływanie się do pełnego sprawozdania z badania poprzedniego rejestrującego.

Artykuł 30 Udostępnianie danych wiążących się z przeprowadzeniem badań • 1. Zanim przeprowadzone zostaną badania w celu uzyskania informacji wymaganych dla celów rejestracji, uczestnik forum SIEF sprawdza poprzez komunikację w ramach swojego forum SIEF, czy odpowiednie wyniki badania są dostępne. Jeżeli odpowiednie wyniki badań na zwierzętach kręgowych są dostępne w obrębie forum SIEF, uczestnik tego forum zwraca się z wnioskiem o udostępnienie wyników tych badań. Jeżeli odpowiednie wyniki badań niewiążących się z badaniami na zwierzętach kręgowych są dostępne w obrębie forum SIEF, uczestnik tego forum może zwrócić się z wnioskiem o udostępnienie wyników tych badań. • Właściciel wyników badania w ciągu miesiąca od złożenia powyższego wniosku przedstawia uczestnikom występującym z tym wnioskiem dokumenty stanowiące dowód kosztów poniesionych na ten cel.

cd. Artykuł 30 Udostępnianie danych wiążących się z przeprowadzeniem badań 3. Jeżeli właściciel wyników badania, o którym mowa w ust. 1, wiążącego się z badaniami na zwierzętach kręgowych, odmawia przedstawienia dokumentów stanowiących dowód kosztów tego badania lub też odmawia udostępnienia samej dokumentacji tego badania innym uczestnikom, nie może kontynuować rejestracji do momentu przedstawienia wspomnianych informacji pozostałym uczestnikom. Pozostali uczestnicy kontynuują rejestrację bez spełnienia wymagania dotyczącego tych informacji, wyjaśniając przyczynę takiego stanu rzeczy w dokumentacji rejestracyjnej. Badania nie powtarza się, chyba że w ciągu 12 miesięcy od daty rejestracji dokonanej przez pozostałych uczestników właściciel informacji nie przekazał ich innym uczestnikom i Agencja podejmie decyzję o powtórzeniu przez nich badań. Jeżeli jednak inny rejestrujący przedłożył już dokumenty rejestracyjne, Agencja udziela pozostałym potencjalnym rejestrującym pozwolenia na odwoływanie się do tych informacji w ich dokumentacjach rejestracyjnych. Wspomnianemu innemu rejestrującemu przysługuje roszczenie o zwrot przez innych uczestników równej części kosztów, którego dochodzić może przed sądami krajowymi, pod warunkiem że udostępnia on innym uczestnikom pełny raport badawczy.

TYTUŁ VI OCENA ROZDZIAŁ 1 Ocena dokumentacji Artykuł 40 Analiza propozycji przeprowadzenia badań

Artykuł 40 Analiza propozycji przeprowadzenia badań 1. Agencja analizuje każdą propozycję przeprowadzenia badań przedstawioną w dokumentach rejestracyjnych lub w sprawozdaniu dalszego użytkownika mającą na celu dostarczenie informacji dotyczących danej substancji określonych w załącznikach IX i X. • Pierwszeństwo przyznaje się dokumentom rejestracyjnym tych substancji, które posiadają lub mogą posiadać właściwości substancji trwałych, wykazujących zdolność do bioakumulacji i toksycznych (PBT) lub bardzo trwałych i wykazujących bardzo dużą zdolność do bioakumulacji (v. Pv. B), właściwości uczulające lub rakotwórcze, mutagenne lub działające szkodliwie na rozrodczość (CMR), a także substancji w ilości powyżej 100 ton rocznie.

cd. Artykuł 40 Analiza propozycji przeprowadzenia badań 2. Informacje odnoszące się do propozycji przeprowadzenia badań obejmujących badania na zwierzętach kręgowych są publikowane na stronach internetowych Agencji. Agencja publikuje na swoich stronach internetowych nazwę substancji, konkluzje na temat zagrożeń, w odniesieniu do których proponuje się przeprowadzenie badania na kręgowcach, a także termin, w którym wymagane są informacje od stron trzecich. Agencja zwraca się do stron trzecich o przedłożenie, z wykorzystaniem dostarczonego przez nią formularza, naukowo uzasadnionych informacji i badań dotyczących danej substancji i konkluzji na temat zagrożeń, których dotyczy propozycja przeprowadzenia badania, w terminie 45 dni od daty publikacji. Wszystkie otrzymane tego typu naukowo uzasadnione informacje i badania są uwzględniane przez Agencję przygotowywaniu decyzji zgodnie z ust. 3.
 w terminie do")
Artykuł 43 Procedura i terminy analizy propozycji przeprowadzenia badań • c) w terminie do dnia 1 czerwca 2022 r. w odniesieniu do wszystkich dokumentów rejestracyjnych zawierających propozycje przeprowadzenia badań i otrzymanych w terminie do dnia 1 czerwca 2018 r.

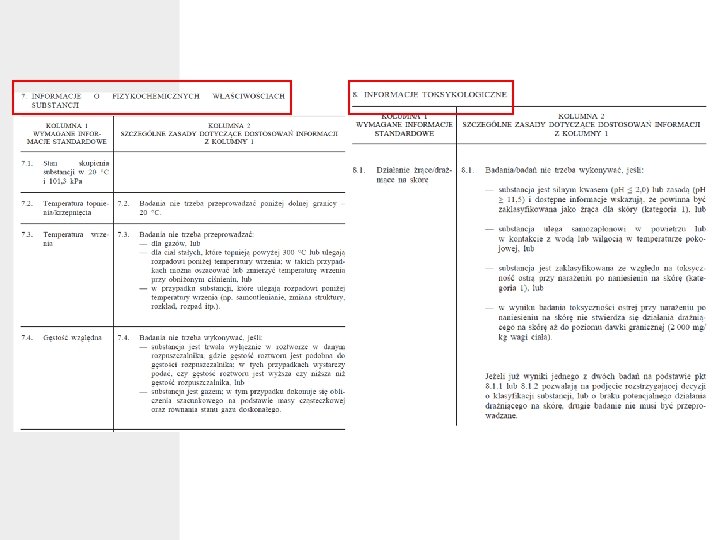
ZAŁĄCZNIK VII WYMAGANIA W ZAKRESIE INFORMACJI STANDARDOWYCH DOTYCZĄCYCH SUBSTANCJI PRODUKOWANYCH LUB IMPORTOWANYCH W ILOŚCI CO NAJMNIEJ 1 TONY W kolumnie 1 niniejszego załącznika określone są standardowe informacje wymagane w odniesieniu do: a) substancji niewprowadzonych produkowanych lub importowanych w ilości 1– 10 ton; b) substancji wprowadzonych produkowanych lub importowanych w ilości 1– 10 ton i spełniających kryteria określone w załączniku III zgodnie z art. 12 ust. 1 lit. a) i b); oraz c) substancji produkowanych lub importowanych w ilości co najmniej 10 ton rocznie.

cd. ZAŁĄCZNIK VII • Przedstawić należy wszelkie inne dostępne istotne informacje fizykochemiczne, toksykologiczne i ekotoksykologiczne. Dla substancji niespełniających kryteriów określonych w załączniku III wymagane są wyłącznie wymogi fizykochemiczne, jak określono w sekcji 7 niniejszego załącznika. • W kolumnie 2 niniejszego załącznika wymienione są szczególne zasady, zgodnie z którymi wymagane informacje standardowe można pomijać, zastępować innymi, podawać na innym etapie lub dostosowywać w inny sposób. Jeśli spełnione są warunki pozwalające na wnioskowanie o dostosowanie wymogów dotyczących informacji zgodnie z kolumną 2, rejestrujący wyraźnie wskazuje na ten fakt oraz na przyczyny wnioskowania o każde z dostosowań w odpowiedniej pozycji dokumentacji rejestracyjnej.


ZAŁĄCZNIK XI OGÓLNE ZASADY DOSTOSOWYWANIA STANDARDOWEGO TRYBU BADAŃ OKREŚLONEGO W ZAŁĄCZNIKACH VII–X • Dodatkowo w stosunku do przepisów szczegółowych określonych w kolumnie 2 załączników VII–X rejestrujący może stosować standardowy tryb badań na podstawie przepisów ogólnych określonych w sekcji 1 niniejszego załącznika. W ramach oceny dokumentacji Agencja może dokonać oceny tych dostosowań w stosunku do standardowego trybu badań.

ZAŁĄCZNIK XI 1. BADANIA NIE WYDAJĄ SIĘ KONIECZNE Z NAUKOWEGO PUNKTU WIDZENIA 1. 1. Wykorzystywanie istniejących danych 1. 1. 1. Dane dotyczące właściwości fizykochemicznych pochodzące z doświadczeń przeprowadzanych niezgodnie z zasadami dobrej praktyki laboratoryjnej (DPL) lub metodami badań, o których mowa w art. 13 ust. 3 Dane uważa się za równoważne z danymi wygenerowanymi za pośrednictwem odpowiednich metod badań, o których mowa w art. 13 ust. 3, jeżeli spełnione są następujące warunki: 1) dane są odpowiednie do celów klasyfikacji i oznakowania lub oceny ryzyka; 2) istnieje wystarczająca dokumentacja umożliwiająca ocenę adekwatności badania; oraz 3) dane zachowują ważność dla analizowanych rodzajów działań, a badanie prowadzone jest na akceptowalnym poziomie jakości.

ZAŁĄCZNIK XI 1. 1. 2. Dane dotyczące działania na zdrowie człowieka i właściwości środowiskowych pochodzące z doświadczeń przeprowadzanych niezgodnie z zasadami dobrej praktyki laboratoryjnej (DPL) lub metodami badań, o których mowa w art. 13 ust. 3 Dane uważa się za równoważne z danymi wygenerowanymi za pośrednictwem odpowiednich metod badań, o których mowa w art. 13 ust. 3, jeżeli spełnione są następujące warunki: 1) dane są odpowiednie do celów klasyfikacji i oznakowania lub oceny ryzyka; 2) dane są adekwatne, wiarygodne i obejmują kluczowe parametry, które mają być analizowane za pośrednictwem odpowiednich metod badań, o których mowa w art. 13 ust. 3; 3) czas trwania narażenia jest porównywalny lub dłuższy w stosunku do odpowiednich metod badań, o których mowa z art. 13 ust. 3, jeśli czas trwania narażenia jest istotnym parametrem; oraz 4) istnieje właściwa i wiarygodna dokumentacja badania

ZAŁĄCZNIK XI 1. 1. 3. Istniejące dane uzyskane w wyniku badań na ludziach Należy wziąć pod uwagę istniejące dane uzyskane w wyniku badań na ludziach, takie jak badania epidemiologiczne narażonych populacji, dane i badania kliniczne w odniesieniu do narażenia przypadkowego oraz w miejscu pracy. Adekwatność danych dotyczących konkretnego skutku dla zdrowia ludzkiego zależy, miedzy innymi, od rodzaju analizy i badanych parametrów oraz od natężenia i charakteru odpowiedzi, a tym samym możliwości przewidzenia skutku. Kryteria oceny adekwatności danych obejmują: 1) odpowiedni wybór i charakterystykę narażonych grup badanych oraz grup kontrolnych; 2) odpowiednią charakterystykę narażenia; 3) okres obserwacji o długości wystarczającej do pojawienia się choroby;
 odpowiednią metodę obserwacji skutku; 5) odpowiednie uwzględnienie czynników ubocznych oraz")
cd. ZAŁĄCZNIK XI 4) odpowiednią metodę obserwacji skutku; 5) odpowiednie uwzględnienie czynników ubocznych oraz mogących spowodować omyłkę; oraz 6) odpowiedni stopień pewności statystycznej w celu uzasadnienia wyniku. We wszystkich przypadkach należy zapewnić odpowiednią i wiarygodną dokumentację.

ZAŁĄCZNIK XI 1. 2. Ciężar dowodu Dowody pochodzące z kilku niezależnych źródeł informacji mogą być wystarczające do uzasadnienia przypuszczenia/konkluzji, że dana substancja posiada konkretne właściwości niebezpieczne lub też takich właściwości nie posiada, podczas gdy informacje pochodzące tylko z jednego źródła uważane są za niewystarczające do uzasadnienia takiego twierdzenia. Dowody pochodzące z wykorzystania nowo opracowanych metod badań, niewłączonych jeszcze do metod badań, o których mowa w art. 13 ust. 3, lub też z wykorzystania międzynarodowej metody badawczej uznanej przez Komisję lub Agencję za równoważną, mogą okazać się wystarczające do wyciągnięcia wniosku na temat tego, czy dana substancja ma dane właściwości niebezpieczne, czy też ich nie ma.

ZAŁĄCZNIK XI W przypadku zgromadzenia wystarczającej ilości dowodów na istnienie lub brak istnienia danej właściwości niebezpiecznej: — należy zrezygnować z dalszych badań tej właściwości na zwierzętach kręgowych, — można zrezygnować z dalszych badań, które nie są prowadzone na zwierzętach kręgowych. We wszystkich przypadkach należy zapewnić odpowiednią i wiarygodną dokumentację.
SAR) Wyniki uzyskane na podstawie")
ZAŁĄCZNIK XI 1. 3. Jakościowa lub ilościowa zależność struktura-aktywność ((Q)SAR) Wyniki uzyskane na podstawie ważnych jakościowych lub ilościowych modeli zależności struktura-aktywność ((Q)SAR) mogą wskazywać na obecność niebezpiecznej właściwości lub jej brak. Wyniki badań (Q)SAR mogą być wykorzystane zamiast badań, gdy spełnione są następujące warunki: — wyniki pochodzą z modelu (Q)SAR o ustalonej ważności naukowej, — substancja należy do dziedziny zastosowania modelu (Q)SAR, — wyniki są adekwatne do celów klasyfikacji i oznakowania lub oceny ryzyka, oraz — przedstawiona jest właściwa i wiarygodna dokumentacja dotycząca stosowanej metody. Agencja we współpracy z Komisją, państwami członkowskimi oraz innymi zainteresowanymi stronami opracowuje i przekazuje wskazówki dotyczące oceny spełnienia wymienionych warunków przez poszczególne modele QSAR, a także dostarcza odpowiednich przykładów.

ZAŁĄCZNIK XI • 1. 4. Metody in vitro • Wyniki uzyskane poprzez zastosowanie odpowiednich metod in vitro mogą wykazać obecność danej właściwości niebezpiecznej lub mogą mieć znaczenie dla rozumienia mechanistycznego, co może mieć znaczenie dla oceny. W tym kontekście „odpowiednie” oznacza wystarczająco dobrze rozwinięte zgodnie z uznanymi, międzynarodowymi kryteriami opracowywania badań (np. kryteria Europejskiego Centrum Walidacji Alternatywnych Metod Badań (ECVAM) dotyczące kwalifikacji badania do wstępnego procesu walidacji). W zależności od potencjalnego ryzyka może być konieczne natychmiastowe potwierdzenie konieczności przeprowadzenia badań dostarczających więcej informacji, niż przewidziano w załącznikach VII lub VIII, lub wniosek dotyczący potwierdzenia konieczności przeprowadzenia badań dostarczających więcej informacji, niż przewidziano w załącznikach IX lub X dla odpowiedniego zakresu wielkości obrotu.

cd. ZAŁĄCZNIK XI • Jeżeli wyniki uzyskane przy użyciu takich metod in vitro nie wskazują na istnienie danej właściwości niebezpiecznej, należy mimo wszystko przeprowadzić przewidziane badanie dla odpowiedniego zakresu wielkości obrotu w celu potwierdzenia negatywnego wyniku, chyba że badanie jest wymagane zgodnie z załącznikami VII–X lub z innymi przepisami niniejszego załącznika. • Można odstąpić od takiego potwierdzania, jeżeli spełnione są następujące warunki: • 1) wyniki pochodzą z badań in vitro, których wartość potwierdzono naukowo poprzez badanie walidacyjne, zgodnie z ustalonymi na poziomie międzynarodowym zasadami walidacji; • 2) wyniki są adekwatne do celów klasyfikacji i oznakowania lub oceny ryzyka; oraz • 3) przedstawiona jest właściwa i wiarygodna dokumentacja dotycząca stosowanej metody.

ZAŁĄCZNIK XI 1. 5. Grupowanie substancji i podejście przekrojowe Substancje, w przypadku których istnieje prawdopodobieństwo podobnych właściwości fizykochemicznych, toksykologicznych oraz ekotoksykologicznych lub zbliżonych ze względu na podobieństwo strukturalne mogą być traktowane jako grupa lub „kategoria” substancji. Zastosowanie pojęcia „grupy” wymaga, aby właściwości fizykochemiczne, skutki działania na zdrowie człowieka oraz skutki działania na środowisko lub losy w środowisku mogły być przewidywane na podstawie danych dotyczących substancji referencyjnej z danej grupy poprzez interpolację względem innych substancji w grupie (podejście przekrojowe). Pozwala to na uniknięcie konieczności badania każdej substancji ze zględu na każdy rodzaj działania. Po konsultacji z zainteresowanymi podmiotami i innymi zainteresowanymi stronami Agencja wydaje wytyczne dotyczące technicznie i naukowo uzasadnionej metodologii grupowania substancji, przed upływem terminu pierwszej rejestracji substancji wprowadzonych.

ZAŁĄCZNIK XI 2. BADANIE JEST TECHNICZNIE MOŻLIWE • Można odstąpić od badania danego rodzaju działania, jeżeli na skutek właściwości danej substancji nie jest technicznie możliwe prowadzenie badania: np. nie można użyć substancji o wysokiej lotności, wysoce reaktywnych lub nietrwałych, gdy mieszanie substancji z wodą może spowodować niebezpieczeństwo pożaru lub wybuchu lub też gdy wymagane przy niektórych badaniach znaczenie substancji pierwiastkiem promieniotwórczym może okazać się niemożliwe. Należy zawsze stosować się do wskazówek zamieszczonych w metodach badań, o których mowa w art. 13 ust. 3, zwłaszcza gdy chodzi o ograniczenia techniczne danej metody.

CLP

Znaczenie badań w kontekście CLP • W celu sklasyfikowania i oznakowania substancji albo mieszaniny jej wytwórcy, importerzy lub dalsi użytkownicy muszą zebrać i ocenić wszelkie dostępne informacje związane z niebezpiecznymi właściwościami danej substancji lub mieszaniny. • Jeśli żadne dane nie są dostępne, należy przeprowadzić badania ekotoksykologiczne i toksykologiczne spełniające wymogi rozporządzenia REACH, z poszanowaniem zasad dobrej praktyki laboratoryjnej OECD i wszelkimi metodami uznanymi na poziomie międzynarodowym i zatwierdzonymi zgodnie z procedurami międzynarodowymi, aby zapewnić wysoką jakość i wiarygodność danych. • Jeśli przeprowadzane są badania na zwierzętach, muszą spełniać wymogi dyrektywy w sprawie ochrony zwierząt laboratoryjnych (dyrektywa 2010/63/EU).

Metody alternatywne wobec badań prowadzonych na zwierzętach • Opracowano wiele metod alternatywnych w celu zastąpienia badań prowadzonych na zwierzętach systemami niewymagającymi wykorzystania zwierząt, ograniczenia liczby zwierząt uczestniczących w badaniach lub udoskonalenia procedur w sposób umożliwiający zmniejszenie bólu i stresu u badanych zwierząt (zasada 3 Rs). • ECHA, podmioty zainteresowane oraz wiele innych organów regulacyjnych zaaprobowały zasadę 3 Rs. Dyrektywa 2010/63/UE w sprawie ochrony zwierząt wykorzystywanych do celów naukowych zawiera wyraźne odniesienie do zasady 3 Rs. • W metodach alternatywnych można także uwzględnić własności chemiczne, prognozy i modele (Q)SAR oraz badania in vitro komórek lub tkanek z zastosowaniem nowych technologii, takich jak genomika i proteomika. Ponadto, można prognozować właściwości toksykologiczne substancji na podstawie informacji z badań substancji analogicznych, opierając się na podejściu przekrojowym, lub badań grupy substancji, opierając się na podejściu kategoryzacyjnym. W celu potwierdzenia tych prognoz należy przedstawić dostateczne informacje oraz właściwe uzasadnienie. • ECHA wspiera metody alternatywne wobec badań na zwierzętach, które spełniają wymagania regulacyjne umożliwiając ocenę zagrożenia, jakie substancje stanowią dla zdrowia ludzkiego i środowiska, a jednocześnie unikając niepotrzebnych badań na zwierzętach.

Przeprowadzanie badań dotyczących zagrożeń dla zdrowia ludzkiego i dla środowiska • Według rozporządzenia CLP zazwyczaj do celów klasyfikacji i oznakowania nie jest wymagane przeprowadzenie nowych badań dotyczących zagrożeń, jakie substancje lub mieszaniny stanowią dla zdrowia i środowiska. • Tylko w przypadku, gdy wyczerpane zostały wszystkie inne możliwości wygenerowania informacji i nie są dostępne żadne dane o dostatecznej wiarygodności i jakości, w celu uzyskania nowych informacji można przeprowadzić badania metodami określonymi w art. 13 ust. 3 rozporządzenia REACH. Zabrania się przeprowadzania badań na ludziach i naczelnych innych niż ludzie. • Przed rozważeniem przeprowadzenia badań in vivo należy ocenić następujące informacje: • dane z badań, • czy badania przeprowadzono z poszanowaniem zasad dobrej praktyki laboratoryjnej, • dane uzyskane w przeszłości w wyniku badań na ludziach, • prognozy (Q)SAR, • dane z badań in vitro oraz • wykorzystanie stosownych informacji dotyczących substancji lub mieszanin analogicznych w celu prognozowania niebezpiecznych właściwości rozpatrywanych substancji lub mieszanin „docelowych” (podejście przekrojowe).

cd. Przeprowadzanie badań dotyczących zagrożeń dla zdrowia ludzkiego i dla środowiska • W przypadku zagrożeń dla zdrowia ludzkiego żadne badania in vitro ani prognozy Q(SAR) nie mogą obecnie w pełni zastąpić badań toksykologicznych prowadzonych w celu określenia wpływu substancji chemicznych na zdrowie w wielu punktach końcowych, w tym także po długotrwałym narażeniu lub po narażeniu przez kilka pokoleń. • Należy zebrać dane dotyczące stosownych dróg narażenia (doustnej, przezskórnej i wziewnej) oraz postaci lub stanów fizycznych, w jakich substancja jest wprowadzana do obrotu i w jakich może być stosowana zgodnie z racjonalnymi oczekiwaniami. • Gdy dostępne dane z badań są jedynie pomocnicze, sprzeczne, niejednoznaczne lub nie można ich łatwo porównać z kryteriami CLP, należy zastosować podejście oparte na ciężarze dowodów z uwzględnieniem oceny ekspertów. Istnieje możliwość wykorzystania w celu klasyfikacji i oznakowania stosownych danych pochodzących z innych źródeł (np. sprawdzonych naukowo badań klinicznych lub epidemiologicznych).

cd. Przeprowadzanie badań dotyczących zagrożeń dla zdrowia ludzkiego i dla środowiska • W przypadku zagrożeń dla środowiska dane wykorzystane w klasyfikacji (narażenie ostre i przewlekłe na zagrożenie środowiska wodnego) opierają się głównie na ustalonych punktach końcowych dotyczących niebezpieczeństwa zatrucia na trzech różnych poziomach troficznych środowiska wodnego przy użyciu znormalizowanych w wysokim stopniu protokołów badań. Ryby, skorupiaki i glony (lub inne rośliny) wykorzystywane są jako odpowiedniki reprezentatywne dla szeregu gatunków i grup taksonomicznych w obrębie każdego poziomu troficznego. W celu określenia toksyczności przewlekłej i współczynników M wykorzystuje się informacje dotyczące losu danej substancji lub mieszaniny w środowisku (rozkład i bioakumulacja) łącznie z danymi dotyczącymi toksyczności.

Przeprowadzanie badań dotyczących zagrożeń fizycznych • Jeśli chodzi o zagrożenia fizyczne, wytwórca, importer lub dalszy użytkownik danej substancji lub mieszaniny mają obowiązek opracować nowe dane, chyba że dostępne są już stosowne i wiarygodne informacje. • Aby określić zagrożenie fizyczne, należy ocenić istniejące dane pod kątem ich przydatności oraz jakości przeprowadzonych badań. • Nowe badania muszą być przeprowadzone zgodnie z uznanym systemem jakości lub przez akredytowane laboratoria (np. zachowujące zgodność normą EN ISO/IEC 17025) w oparciu o metody lub normy, o których mowa w części 2 załącznika I do rozporządzenia CLP. • Badania te należy przeprowadzić z użyciem substancji i mieszaniny w odpowiednim stanie fizycznym i w formie, w jakiej zostały wprowadzone do obrotu, Jeżeli na przykład do celów dostawy lub transportu ten sam materiał chemiczny ma występować w postaci fizycznej różnej od tej, w której był badany, i jeżeli uważa się, że może on w sposób istotny zmienić swoje zachowanie w badaniu klasyfikacyjnym, materiał ten należy przebadać również w nowej postaci. • Takie parametry, jak stężenie, kształt, wielkość cząsteczki, gęstość, itp. mogą również wpływać na wyniki badań, a zatem należy je podać.

Artykuł 7 Badania na zwierzętach i na ludziach 1. W przypadku przeprowadzania nowych badań dla celów niniejszego rozporządzenia badania na zwierzętach w rozumieniu dyrektywy 86/609/EWG przeprowadza się wyłącznie wówczas, gdy nie ma innego rozwiązania, które zagwarantowałoby odpowiednią wiarygodność i jakość danych. 2. Do celów niniejszego rozporządzenia zabrania się przeprowadzania badań na naczelnych innych niż ludzie. 3. Do celów niniejszego rozporządzenia zabrania się przeprowadzania badań na ludziach. Do celów niniejszego rozporządzenia można jednak wykorzystywać dane uzyskane z innych źródeł, np. z badań klinicznych.

Artykuł 8 Generowanie nowych informacji dotyczących substancji i mieszanin 1. Aby stwierdzić, czy dana substancja lub mieszanina stwarza zagrożenie dla zdrowia ludzi lub środowiska, o czym mowa w załączniku I do niniejszego rozporządzenia, producent, importer lub dalszy użytkownik mogą przeprowadzić nowe badania, pod warunkiem że wyczerpali już wszelkie inne możliwości wygenerowania informacji, w tym również przez zastosowanie zasad przewidzianych w sekcji 1 załącznika XI do rozporządzenia (WE) nr 1907/2006. 2. Aby stwierdzić, czy substancja lub mieszanina stwarza którekolwiek z zagrożeń wynikających z właściwości fizycznych, o których mowa w części 2 załącznika I, producent, importer lub dalszy użytkownik wykonują badania wymagane zgodnie z tą częścią, chyba że są już dostępne odpowiednie i wiarygodne informacje.

cd. Artykuł 8 Generowanie nowych informacji dotyczących substancji i mieszanin 3. Badania, o których mowa w ust. 1, prowadzone są zgodnie z jedną z następujących metod: a) metody badań, o których mowa w art. 13 ust. 3 rozporządzenia (WE) nr 1907/2006; lub b) solidne, uznane w skali międzynarodowej zasady naukowe lub metody zatwierdzone zgodnie z procedurami międzynarodowymi. • 4. W przypadku gdy producent, importer lub dalszy użytkownik wykonują nowe badania i analizy ekotoksykologiczne lub toksykologiczne, są one realizowane zgodnie z art. 13 ust. 4 rozporządzenia (WE) nr 1907/2006.

cd. Artykuł 8 Generowanie nowych informacji dotyczących substancji i mieszanin 5. W przypadku gdy do celów niniejszego rozporządzenia przeprowadza się nowe badania dotyczące zagrożeń wynikających z właściwości fizycznych, najpóźniej od dnia 1 stycznia 2014 r. są one przeprowadzane zgodnie z odpowiednim uznanym systemem jakości lub przez laboratoria spełniające wymagania odpowiednich uznanych norm. 6. Badania substancji lub mieszaniny, przeprowadzane do celów niniejszego rozporządzenia, przeprowadza się w postaci lub stanie(- ach) fizycznym(-ch), w którym(-ch) ta substancja lub mieszanina jest wprowadzana do obrotu i w których może być stosowana zgodnie z racjonalnymi oczekiwaniami.

Wytyczne OECD i UE dotyczące badań • Badania dotyczące wymagań informacyjnych REACH dotyczących ekotoksyczności, toksyczności i właściwości fizykochemicznych są zazwyczaj opracowywane z wykorzystaniem wytycznych dotyczących badań. Te oficjalne wytyczne zostały zatwierdzone przez OECD i UE. • W związku z rozwojem naukowym i regulacyjnym uaktualnia się wytyczne dotyczące badań i wprowadza się nowe wytyczne dotyczące badań. Dzięki tej stronie internetowej ECHA wspiera rejestrujących, wskazując, jak te wytyczne dotyczące badań można wykorzystać do spełnienia wymogów REACH w zakresie informacji. Przykładowo opisana jest odpowiednio rola nowych wytycznych w zakresie strategii badań. Informacje te są udzielane przed oficjalną aktualizacją poradników ECHA.

Badania dotyczące wymagań REACH: https: //echa. europa. eu/pl/support/oecd-eu-testguidelines

Ocena na podstawie rozporządzenia REACH • ECHA i państwa członkowskie oceniają informacje przedłożone przez przedsiębiorstwa, aby przeanalizować jakość dokumentacji rejestracyjnej i propozycji przeprowadzenia badań. Ocena służy ponadto ustaleniu, czy dana substancja stwarza ryzyko dla zdrowia człowieka lub środowiska. • Prawidłowa identyfikacja substancji jest ważnym elementem procesów oceny, ponieważ pozwala ECHA i państwom członkowskim stwierdzić, że każda rejestracja obejmuje tylko jedną substancję oraz że dane badawcze są odpowiednie dla tej substancji. Profil tożsamości substancji (SIP) i zgłaszanie granicznego składu zapewniają transparentność pod względem istotności danych badawczych.

Jak unikać zbędnych badań na zwierzętach • Na mocy REACH badania na kręgowcach (np. szczurach, innych ssakach lub rybach) mogą być wykorzystywane do wypełnienia obowiązków informacyjnych związanych z rejestracją jedynie w ostateczności. Przedsiębiorstwa wytwarzające lub importujące od 1 do 100 ton na rok mają wiele możliwości uniknięcia zbędnych badań na zwierzętach i zmniejszenia liczby takich badań. • W odniesieniu do każdego pojedynczego wymogu informacyjnego należy rozważyć: • gromadzenie istniejących danych i wzajemne ich udostępnianie. Można uzyskać dostęp do opublikowanego piśmiennictwa, który wystarczy do spełnienia wymogu informacyjnego. Jeżeli wynik prawidłowo przeprowadzonego badania na zwierzętach jest dostępny w SIEF, musi zostać udostępniony współrejestrującym. Właściciel badania musi otrzymać wynagrodzenie według wcześniej ustalonych zasad. • uchylenie lub dostosowanie wymogów dotyczących przedstawiania danych: zasady dostosowywania wymogów stanowią część tekstu aktu prawnego. Mogą być albo szczegółowe (w kolumnie 2 każdego punktu końcowego), albo ogólne (na mocy załącznika XI).

cd. Jak unikać zbędnych badań na zwierzętach • W celu skorzystania z zasad ogólnych można uchylić wymóg dotyczący przedstawiania danych lub dostosować ten wymóg, na podstawie następujących argumentów naukowych: • ciężar dowodów. Mają Państwo wystarczająco dużo informacji z kilku niezależnych źródeł, które prowadzą do wniosku, że Państwa substancja posiada określoną właściwość (lub nie). • modele QSAR. Wiele właściwości substancji można przewidzieć na podstawie właściwości substancji o podobnej budowie, z wykorzystaniem modeli komputerowych. • metody in vitro. Badania przeprowadzone na wyizolowanych tkankach, narządach lub komórkach zamiast całego organizmu mogą wystarczyć do wyciągnięcia wniosków dotyczących wymogu informacyjnego. • grupowanie i podejście przekrojowe. Gdy substancja jest (strukturalnie) podobna do innej substancji (lub grupy substancji), istniejące wyniki dotyczące tych innych substancji mogą być wykorzystane w ramach podejścia przekrojowego w odniesieniu do Państwa substancji. • Decyzja o skorzystaniu z jednej z tych możliwości oznacza ubieganie się o dostosowanie wymogów.

Cd. Badania na zwierzętach na mocy REACH • Głównymi środkami zapewnianymi przez rozporządzenie REACH w celu ograniczenia liczby badań na zwierzętach do niezbędnego minimum są: • Udostępnianie danych • Alternatywne metody i podejścia

Cd. Badania na zwierzętach na mocy REACH Udostępnianie danych • Przedsiębiorstwa produkujące lub przywożące tę samą substancję muszą współpracować i dzielić się informacjami na temat właściwości swoistych ich substancji. Przedsiębiorstwa rejestrujące te same chemikalia muszą dzielić się wynikami badań na zwierzętach kręgowych i wspólnie przekazywać wszystkie informacje do ECHA. Nie można powtarzać odpowiednich i wiarygodnych badań na zwierzętach. Przedsiębiorstwa zachęca się również do dzielenia się wszelkimi dostępnymi informacjami, które posiadają.

Analiza propozycji przeprowadzenia badań • Badania na kręgowcach są wykorzystywanym w ostateczności sposobem uzyskania brakujących informacji dotyczących substancji oraz spełnienia wymagań w zakresie informacji w ramach systemu REACH. ECHA bada wszystkie propozycje w celu sprawdzenia, czy wygenerowane zostaną wiarygodne i odpowiednie dane, jak również w celu zapobieżenia zbędnym badaniom na zwierzętach. • ECHA publikuje każdą propozycję przeprowadzenia badań, która obejmuje kręgowce, i wzywa strony trzecie do przekazywania naukowo uzasadnionych informacji lub badań dotyczących przedmiotowej substancji oraz właściwości krytycznych, które mogą zostać uwzględnione przez ECHA podczas przygotowywania przez nią decyzji w sprawie propozycji przeprowadzenia badań. Informacje należy przekazać w terminie 45 dni. • W przypadku projektu decyzji istnieją następujące opcje: • Przyjęcie propozycji przeprowadzenia badań przy uwzględnieniu zmian warunków badania • Przyjęcie lub odrzucenie propozycji przeprowadzenia badań, jednak w połączeniu z wymogiem przeprowadzenia co najmniej jednego dodatkowego badania • Odrzucenie propozycji przeprowadzenia badań • ECHA przyjmuje decyzję na podstawie wniosku oraz informacji przekazanych przez strony trzecie. • W przypadku gdy decyzja uwzględnia którąkolwiek z trzech pierwszych opcji, a w odniesieniu do tej samej substancji przedłożono kilka propozycji przeprowadzenia badań, rejestrujący muszą uzgodnić, kto przeprowadzi badanie.

cd. Analiza propozycji przeprowadzenia badań Niedopuszczalne propozycje przeprowadzenia badań • Istnieje wiele przyczyn zakończenia analizy propozycji przeprowadzenia badań przed skierowaniem sprawy do właściwych organów państwa członkowskiego. Należą do nich: przerwanie produkcji lub importu przez rejestrującego, wycofanie propozycji przeprowadzenia badań, a także niedopuszczalność. • Niedopuszczalne propozycje przeprowadzenia badań to takie propozycje, w przypadku których REACH nie przewiduje analizy propozycji przeprowadzenia badań. Przypadki te mają miejsce, gdy: • propozycja uwzględnia parametry docelowe określone w załączniku VII i VIII; • badania są już w toku lub zostały zakończone; • przedłożono propozycję przeprowadzenia badań zamiast wyników badań w celu uwzględnienia poprzedniej decyzji właściwego organu państwa członkowskiego zgodnie z art. 16 ust. 1 lub 2 dyrektywy dotyczącej klasyfikacji, pakowania i oznakowania substancji niebezpiecznych (zob. także art. 135 REACH).

Określanie wymagań w zakresie informacji standardowych • Każdy rejestrujący musi określić swoje wymagania w zakresie informacji zgodnie z ilościami produkowanej lub importowanej substancji na podstawie załączników VII–X do rozporządzenia REACH. • Standardowe wymagania mogą się różnić w zależności od tego, czy następujące kryteria mają zastosowanie do substancji: • (i) szczególne przypadki w celu rejestracji substancji od 1 do 10 ton rocznie: jeżeli mogą Państwo wykazać, że przewidywane ryzyko stosowania Państwa substancji jest niskie, mogą Państwo skorzystać z uproszczonych wymagań w zakresie informacji; • (ii) szczególne kryteria przedstawione w kolumnie 2 w załącznikach VII–X, • (iii) ogólne kryteria dostosowywania wymagań w zakresie informacji, przedstawione w załączniku XI. • Aby spełnić swoje wymagania w zakresie informacji, rejestrujący muszą korzystać z istniejących informacji i metod niebadawczych. Badania na kręgowcach są dopuszczalne wyłącznie w ostateczności. Badania odnoszące się do wymagań w zakresie informacji dotyczących ekotoksyczności, toksyczności i właściwości fizykochemicznych należy opracowywać na podstawie zatwierdzonych przez OECD i UE wytycznych dotyczących badań. • Poniżej znajduje się uproszczony wykaz wymagań w zakresie informacji standardowych dla dwóch najniższych wielkości obrotu, które podlegają ostatecznemu terminowi rejestracji w 2018 r. Pełny wykaz wymagań w zakresie informacji, w tym dotyczących większych wielkości obrotu, znajduje się w Poradniku dotyczącym rejestracji (sekcja 3. 1). • Uwaga: wykazy te mogą ulec zmianie wraz z aktualizacją załączników do rozporządzenia REACH lub opracowaniem nowych metodologii.

Wymagania w zakresie informacji: 1– 10 ton rocznie Parametry docelowe dla bezkręgowców Opis stanu skupienia substancji w temperaturze 20°C i przy ciśnieniu 101, 3 k. Pa Informacje wymagane dla najniższej wielkości obrotu są określone w kolumnie 1 załącznika VII do rozporządzenia REACH i zawierają określone dane fizykochemiczne oraz informacje toksykologiczne i ekotoksykologiczne. https: //echa. europa. eu/pl/regulations/reach/regis tration/information-requirements Temperatura topnienia/krzepnięcia Temperatura wrzenia (o ile dotyczy) Gęstość względna Prężność par (o ile dotyczy) Napięcie powierzchniowe (o ile dotyczy) Rozpuszczalność w wodzie Współczynnik podziału Temperatura zapłonu Palność Właściwości wybuchowe Temperatura samozapłonu Właściwości utleniające Granulometria (o ile dotyczy) Badanie in vitro żrącego/drażniącego działania na skórę Badanie in vitro działania drażniącego na oczy Badanie działania drażniącego na skórę Badanie In vitro mutacji genowych u bakterii Badanie toksyczności krótkookresowej na bezkręgowcach Badanie zahamowania wzrostu na roślinach wodnych Łatwość ulegania biodegradacji (o ile dotyczy) Parametry docelowe dla kręgowców Toksyczność ostra: narażenie przez drogi pokarmowe

Wymagania w zakresie informacji: 10 -100 ton rocznie • Informacje wymagane do zwykłej rejestracji o wielkości obrotu wynoszącej 10 -100 ton rocznie (załącznik VIII do rozporządzenia REACH) Uwaga: poniższe dane należy przedstawić jako uzupełnienie do informacji, o których mowa powyżej • * Można przeprowadzić badanie in vivo tylko, jeśli nie mogą Państwo sklasyfikować swojej substancji na podstawie wyników badań in vitro. • https: //echa. europa. eu/pl/regulations/reach/registration/informationrequirements Parametry docelowe dla bezkręgowców Parametry docelowe dla kręgowców Badanie mutagenności in vitro na komórkach ssaków lub badanie mikrojądrowe invitro Badanie in vivo działania drażniącego na skórę Badanie mutacji genowych na komórkach ssaków in vitro Badanie in vivo działania drażniącego na oczy* Test zahamowania oddychania osadu czynnego Propozycja przeprowadzenia badań genotoksyczności in vivo (o ile dotyczy) Rozkład Toksyczność ostra: narażenie przez drogi oddechowe Hydroliza Badanie krótkookresowej toksyczności dawki powtórzonej (28 dni) Test przesiewowy adsorpcji/desorpcji Przesiewowe badanie szkodliwego działania na rozrodczość/rozwój Krótkookresowa toksyczność dla ryb lub propozycja przeprowadzenia badań przewlekłej toksyczności dla ryb (o ile dotyczy)

Źródla: • https: //echa. europa. eu/pl/regulations/reach/registration/information-requirements • https: //echa. europa. eu/pl/testing-clp • https: //echa. europa. eu/pl/support/registration/how-to-avoid-unnecessary-testing-onanimals/weight-of-evidence • https: //echa. europa. eu/pl/support/oecd-eu-test-guidelines

Czym jest Dobra Praktyka Laboratoryjna DPL ?

Dobra Praktyka Laboratoryjna to system jakości odnoszący się do procesu organizacyjnego i warunków planowania, przeprowadzania i monitorowania nieklinicznych badań substancji i ich mieszanin pod względem bezpieczeństwa dla zdrowia człowieka i środowiska oraz dokumentowania, archiwizowania i prezentowania wyników takich badań. Zasady Dobrej Praktyki Laboratoryjnej stosuje się w nieklinicznych badaniach dotyczących bezpieczeństwa produktów leczniczych, weterynaryjnych produktów leczniczych, środków ochrony roślin, kosmetyków, produktów biobójczych, dodatków do żywności, dodatków do pasz, detergentów oraz chemikaliów stosowanych w przemyśle, usługach i gospodarstwie domowym. Badania, do których stosuje się zasady Dobrej Praktyki Laboratoryjnej, obejmują badania w laboratoriach, badania w szklarniach i badania polowe.
")
Dobra Praktyka Laboratoryjna (DPL)
\" • na początku lat 70 -tych FDA zdała sobie")
Koncepcja "Dobrej Praktyki Laboratoryjnej (DPL)" • na początku lat 70 -tych FDA zdała sobie sprawę z przypadków (PLP) złej praktyki laboratoryjnej na terenie całych Stanów Zjednoczonych; • przeprowadzono szczegółowe dochodzenie w 40 laboratoriach toksykologicznych: • nie skalibrowana aparatura pomiarowa; • nieprawidłowe / niedokładne obliczenia; • niewłaściwe procedury badawcze; • w jednym z największych laboratoriów Industrial Bio-Test Laboratories, (badania dla dużych firm, takich jak Procter and Gamble czy Monsanto), stwierdzono że u myszy, które zostały użyte w badaniach toksykologicznych płynu kosmetycznego i dezodorantu rozwinął się rak w skutek czego zmarło 89 zwierząt, Bio test usunął martwe myszy z badań i sfabrykował wyniki uznając produkty za bezpieczne dla ludzi. • powstała w USA w 1970 roku. FDA proponuje rozporządzenia dotyczące GLP w 1976 roku, a ostateczne ustalenia reguły w czerwcu 1979 r. (Code of Federal Regulations 21 CFR 58). • w 1981 roku OECD ogłosiło zasady DPL, które stają się międzynarodowym standardem.

 Została")
Organizacja Współpracy Gospodarczej i Rozwoju (Organisation for Economic Cooperation and Development – OECD) Została utworzona na mocy podpisanej 14 grudnia 1960 r Konwencji Paryskiej i zastąpiła powstałą 16 kwietnia 1948 r. Organizację Europejskiej Współpracy Gospodarczej (OEEC), której zadaniem było integrowanie gospodarek państw Europejskich, korzystających z pomocy w ramach Planu Marshalla. Początkowo w skład OECD wchodziło 17 państw Europy Zachodniej oraz Stany Zjednoczone, Kanada i Turcja. Obecnie w skład Organizacji wchodzą 34 najbardziej rozwinięte gospodarczo państwa świata o ustroju demokratycznym. Polska jest pełnoprawnym członkiem OECD od 22 listopada 1996 r. Opracowane przez OECD zasady DPL zostały po raz pierwszy implementowane do systemu prawnego Unii Europejskiej na mocy Dyrektywy Komisji 87/18/EWG z dnia 18 grudnia 1986 r. i znowelizowane Dyrektywą Komisji 1999/11/WE z dnia 8 marca 1999 r. Tekst jednolity zawarto w dyrektywie Parlamentu Europejskiego i Komisji 2004/10/WE z dnia 11 lutego 2004. • Siedzibą OECD jest Château de la Muette w Paryżu.
 Dokumenty")
Organizacja Współpracy Gospodarczej i Rozwoju (Organisation for Economic Cooperation and Development – OECD) Dokumenty OECD dotyczące Dobrej Praktyki Laboratoryjnej w tym akty Rady oraz wytyczne można znaleźć w Internecie na stronie: http: //www. oecd. org/chemicalsafety/testing/oecdseriesonprinc iplesofgoodlaboratorypracticeglpandcompliancemonitoring. htm Metody badawcze: http: //www. oecd. org/env/ehs/testing/oecdguidelinesforthetesti ngofchemicals. htm

Unia Europejska, UE • http: //ec. europa. eu/growth/sectors/chemicals/good-laboratorypractice_en • Akty prawne UE dotyczące DPL • Rozporządzenie Komisji (WE) Nr 440/2008 z dnia 30 maja 2008 r. ustalające metody badań zgodnie z rozporządzeniem (WE) nr 1907/2006 Parlamentu Europejskiego i Rady w sprawie rejestracji, oceny, udzielania zezwoleń i stosowanych ograniczeń w zakresie chemikaliów (REACH) Dz. Urz. UE L 142 z 31. 05. 2008 • Dyrektywa 2004/9/WE Parlamentu Europejskiego i Rady z dnia 11 lutego 2004 r. w sprawie kontroli i weryfikacji dobrej praktyki laboratoryjnej (DPL) (wersja skodyfikowana). Dz. Urz. UE L 50 z 20. 02. 2004 • Dyrektywa 2004/10/WE Parlamentu Europejskiego i Rady z dnia 11 lutego 2004 r. w sprawie harmonizacji przepisów ustawowych, wykonawczych i administracyjnych odnoszących się do stosowania zasad dobrej praktyki laboratoryjnej i weryfikacji jej stosowania na potrzeby badań substancji chemicznych (wersja skodyfikowana) - Dz. Urz. UE L 50 z 20. 02. 2004
 dane uzyskiwane w badaniach substancji chemicznych")
Wzajemne akceptowanie danych (Mutual Acceptance of Data (MAD) dane uzyskiwane w badaniach substancji chemicznych przeprowadzonych w kraju członkowskim OECD w zgodności z Wytycznymi OECD do Badań Substancji Chemicznych oraz Zasadami Dobrej Praktyki Laboratoryjnej OECD będą uznawane w innych krajach członkowskich, dla celów oceny oraz innych zastosowań związanych z ochroną człowieka i środowiska”.
 https: //it-consulting. pl/autoinstalator/wordpress /2015/10/02/ekonomia-to-takze-teoria-systemow/ •")
cd. Wzajemne akceptowanie danych (Mutual Acceptance of Data (MAD) https: //it-consulting. pl/autoinstalator/wordpress /2015/10/02/ekonomia-to-takze-teoria-systemow/ • ujednolicenie wymaganych prawem nieklinicznych badań laboratoryjnych substancji i ich mieszanin pod względem bezpieczeństwa dla zdrowia człowieka i środowiska oraz dokumentowania, archiwizowania i prezentowania wyników takich badań; • zapewnienie wysokiej jakości i wiarygodności wyników badań; • uznanie procedur monitorowania zgodności z Zasadami Dobrej Praktyki Laboratoryjnej ułatwia wzajemne uznawanie danych i przyczyna się w ten sposób do zredukowania powielania badań substancji chemicznych; • zmniejszenia powielania badań substancji chemicznych, a co za tym idzie zmniejszenie liczby zwierząt wykorzystywanych w badaniach. • ekonomiczne – oszczędza się czas i zasoby finansowe

Metody badawcze • Oprócz stosowania zasad DPL i monitorowania ich spełniania, istotnym elementem umożliwiającym wzajemną akceptację wyników badań są wiarygodne, ujednolicone metody badań. • W Unii Europejskiej i w Polsce metody badań określone są w wielokrotnie nowelizowanym rozporządzeniu Komisji (WE) nr 440/2008 z dnia 30 maja 2008 r. ustalającym metody badań zgodnie z rozporządzeniem (WE) nr 1907/2006 Parlamentu Europejskiego i Rady w sprawie rejestracji, oceny, udzielania zezwoleń i stosowanych ograniczeń w zakresie chemikaliów (REACH).

OECD Test Guidelines for the Chemicals Sekcja 1: Physical Chemical Properties (Zakres certyfikatu DPL – 1: badania właściwości fizykochemicznych) Sekcja 2: Effects on Biotic Systems (Zakres certyfikatu DPL – 4: badania właściwości toksyczności środowiskowej w odniesieniu do organizmów wodnych i lądowych oraz 7 badania wpływu na układy typu mezokosm i ekosystemy naturalne) Sekcja 3: Environmental Fate and Behaviour (Zakres certyfikatu DPL – 5: badania zachowania się badanej substancji w wodzie, glebie i powietrzu, badania biakumulacji oraz) Sekcja 4: Health Effects (Zakres certyfikatu DPL – 2: badania właściwości toksycznych oraz 3 badania właściwości mutagennych) Sekcja 5: Other Test Guidelines (Zakres certyfikatu DPL – 6: badania pozostałości)

Inne metody badawcze § metody CIPAC www. cipac. org - International Pesticides Analytical Council (np. Przyspieszone starzenie wg CIPAC MT 46. 3 Accelerated Storage Procedure; p. H wg CIPAC MT 75. 3 Determination of p. H values) § metody ISO (np PN-EN ISO 10993 -10 „Biologiczna ocena wyrobów medycznych. Część 10. Badanie działania drażniącego i uczulającego na skórę) § Farmakopea Polska (np. wygląd, barwa i zapach) § US EPA – np. wygląd, barwa i zapach (OPPTS 830. 6302 Color, EPA OPPTS 830. 6303 Physical State, EPA OPPTS 830. 6304 Odor) § Manual of Tests and Criteria (Podręcznik badań i kryteriów) § metody własne zwalidowane

Zakres wykonywanych badań 1. Badania właściwości fizykochemicznych; 2. Badania właściwości toksycznych; 3. Badania właściwości mutagennych; 4. Badania toksyczności środowiskowej w odniesieniu do organizmów wodnych i lądowych; 5. Badania zachowania się badanej substancji w wodzie, glebie i powietrzu, badania bioakumulacji; 6. Badania pozostałości; 7. Badania wpływu na układy typu mezokosm i ekosystemy naturalne; 8. Analizy chemiczne i badania biochemiczne. 9. Inne badania

Kontrola i weryfikacja spełniania przez jednostki badawcze zasad DPL nie dotyczy merytorycznej oceny celu, przedmiotu i metodyki badań. Koncentruje się natomiast na: • zasobach(organizacja jednostki badawczej, personel, pomieszczenia i wyposażenie); • regułach przeprowadzania badań (plany badań, Standardowe Procedury Operacyjne, funkcja, zadania i odpowiedzialność kierownika badania); • dokumentacji(dane źródłowe, sprawozdanie z badania, obieg dokumentów, archiwizacja przechowywanie w archiwum); • mechanizmie zapewnienia przestrzegania zasad DPL w jednostce opisane w Programie zapewnienia jakości.

Terminologia stosowana w zasadach DPL http: //inmyownterms. com/wp-content/uploads/2015/04/use-mouse-to-open-window-pic. gif 81
 § jednostka organizacyjna wykonująca badania substancji lub ich mieszanin, posiadająca")
JEDNOSTKA BADAWCZA (TEST FACILITY) § jednostka organizacyjna wykonująca badania substancji lub ich mieszanin, posiadająca certyfikat DPL i wpisanych do wykazu jednostek badawczych certyfikowanych w zakresie DPL § kadra, warunki lokalowe i wyposażenie niezbędne do przeprowadzania nieklinicznych badań z zakresu bezpieczeństwa i zdrowia człowieka i środowiska MIEJSCE BADANIA (TEST SITE) § miejsce, w którym jest wykonywany dany etap lub etapy badania; Terminologia stosowana w zasadach DPL 82
 § odpowiada za organizację i działanie jednostki badawczej")
ZARZĄDZAJĄCY JEDNOSTKĄ BADAWCZĄ (TEST FACILITY MANAGEMENT) § odpowiada za organizację i działanie jednostki badawczej zgodnie z zasadami Dobrej Praktyki Laboratoryjnej ZARZĄDZAJĄCY MIEJSCEM BADANIA (TEST SITE MENEGMENT) § osoba lub osoby odpowiedzialne za zagwarantowanie, że dany etap lub etapy badania, za który są odpowiedzialne, są wykonywane zgodnie z zasadami Dobrej Praktyki Laboratoryjnej; ZLECENIODAWCA (SPONSOR) § podmiot zlecający niekliniczne badanie substancji chemicznych i ich mieszanin z zakresu bezpieczeństwa dla zdrowia człowieka i środowiska; Terminologia stosowana w zasadach DPL 83
 § osoba odpowiedzialna za całość przeprowadzanego nieklinicznego badania z zakresu")
KIEROWNIK BADANIA (STUDY DIRECTOR) § osoba odpowiedzialna za całość przeprowadzanego nieklinicznego badania z zakresu bezpieczeństwa i zdrowia człowieka i środowiska GŁÓWNY WYKONAWCA (PRINCIPAL INVESTIGATOR) § osoba, która w przypadku badań wykonywanych w wielu miejscach jest upoważniona przez kierownika badania do działania w jego imieniu jako odpowiedzialna za wydzielony etap badania; upoważnienie głównego wykonawcy do działania w imieniu kierownika badania nie obejmuje odpowiedzialności za całokształt badań i działań, takich jak: zatwierdzanie planu badania i jego zmian, zatwierdzanie raportu końcowego i zapewnienie, że wszystkie mające zastosowanie zasady Dobrej Praktyki Laboratoryjnej są przestrzegane Terminologia stosowana w zasadach DPL 84
 § procedury wykonywane przez personel niezależny od przeprowadzanego")
PROGRAM ZAPEWNIENIA JAKOŚCI (QUALITY ASSURANCE PROGRAMME) § procedury wykonywane przez personel niezależny od przeprowadzanego badania, wprowadzone w jednostce badawczej w celu zapewnienia zarządzającemu jednostką badawczą zgodności przeprowadzanych badań z zasadami Dobrej Praktyki Laboratoryjnej; Terminologia stosowana w zasadach DPL 85
 § udokumentowane procedury określające sposób przeprowadzenia badania, a")
STANDARDOWE PROCEDURY OPERACYJNE (STANDARD PROCEDURE OPERATING) § udokumentowane procedury określające sposób przeprowadzenia badania, a także określające postępowanie nieopisane dokładnie w planach badania lub w wytycznych do badań; PLAN DZIAŁANIA JEDNOSTKI BADAWCZEJ (MASTER SCHEDULE) § zbiór informacji pozwalających na ocenę obciążenia pracą w jednostce badawczej oraz na monitorowanie przebiegu badań wykonywanych w takiej jednostce; Terminologia stosowana w zasadach DPL 86
 § badanie trwające przez krótki okres, z zastosowaniem powszechnie stosowanych,")
BADANIE KRÓTKOTERMINOWE (SHORT-TERM STUDY) § badanie trwające przez krótki okres, z zastosowaniem powszechnie stosowanych, rutynowych metod badań Terminologia stosowana w zasadach DPL 87
 § dokument wraz z wszelkimi poprawkami, określający cele i planowany")
PLAN BADANIA (STUDY PLAN) § dokument wraz z wszelkimi poprawkami, określający cele i planowany przebieg badania POPRAWKA DO PLANU BADANIA (STUDY PLAN AMENDMENT) § celowo wprowadzone po dacie uzasadnione zmiany w planie badania rozpoczęcia badania ODSTĘPSTWO OD PLANU BADANIA (STUDY PLAN DEVIATION) § niezamierzoną zmianę w planie badania po dniu rozpoczęcia badania; Terminologia stosowana w zasadach DPL 88
 § system biologiczny, chemiczny lub fizyczny albo ich połączenie, zastosowane")
SYSTEM BADAWCZY (TEST SYSYTEM) § system biologiczny, chemiczny lub fizyczny albo ich połączenie, zastosowane w badaniach PRÓBKA (SPECIMEN) § każdy materiał pochodzący z systemu badawczego, pobrany w celu zbadania, analizy lub w celu przechowania Terminologia stosowana w zasadach DPL 89
 § dzień, w którym kierownik badania podpisał plan")
DATA ROZPOCZĘCIA BADANIA (STUDY INITIATION DATE) § dzień, w którym kierownik badania podpisał plan badania DATA ROZPOCZĘCIA CZĘŚCI EXPERYMENTLANEJ BADANIA (EXPERIMANTAL STARTING DATE) § dzień, w którym uzyskano pierwsze informacje specyficzne dla tego badania DATA ZAKOŃCZENIA CZĘŚCI EXPERYMENTLANEJ BADANIA (EXPERIMANTAL COMLETION DATE) § dzień, w którym uzyskano ostatnie dane DATA ZAKOŃCZENIA BADANIA (STUDY COMPLETION DATE) § dzień, w którym kierownik badania podpisał sprawozdanie końcowe Terminologia stosowana w zasadach DPL 90
 § materiał będący przedmiotem badania; MATERIAŁ ODNIESIENIA (REFERENCE ITEM “CONTROL")
MATERIAŁ BADANY (TEST ITEM) § materiał będący przedmiotem badania; MATERIAŁ ODNIESIENIA (REFERENCE ITEM “CONTROL ITEM”) § każdy właściwie scharakteryzowany materiał użyty w celach porównawczych SERIA (BATCH) § określona ilość lub partia materiału badanego lub materiału odniesienia, wytworzonego podczas określonego cyklu wytwarzania w taki sposób, że można oczekiwać, że ma ona jednolity charakter NOŚNIK (VEHICLE) § każdy środek zastosowany w celu wymieszania, rozproszenia lub rozpuszczenia materiału badanego lub materiału odniesienia, w celu ułatwienia jego wprowadzenia lub podania do systemu badawczego. Terminologia stosowana w zasadach DPL 91
 § z zakresu bezpieczeństwa i zdrowia")
BADANIE NIEKLINICZNE (NON-CLINICAL HEALTH AND ENVIRONMENTAL SAFETY STUDY) § z zakresu bezpieczeństwa i zdrowia człowieka, i środowiska oznacza doświadczenie lub zespół doświadczeń, w których materiał badany poddawany jest w warunkach laboratoryjnych lub w środowisku badaniom w celu uzyskania danych na temat jego właściwości i/lub bezpieczeństwa stosowania, i których wyniki będą przedłożone właściwym organom Terminologia stosowana w zasadach DPL 92

Terminologia stosowana w zasadach DPL 93

Organizacja jednostki badawczej i jej personel ZARZĄDZAJĄCY JEDNOSTKĄ BADAWCZĄ ARCHIWISTA KIEROWNIK BADANIA JZJ/QA PROGRAM ZAPEWNIENIA JAKOŚCI

Program zapewnienia jakości • Jednostka badawcza posiada program zapewnienia jakości, sporządzony w formie pisemnej, gwarantujący prowadzenie badań zgodnie z zasadami Dobrej Praktyki Laboratoryjnej. • JZJ odpowiada za podejmowanie działań mających zagwarantować prowadzenie badań zgodnie z zasadami DPL tj: • zarządzanie kopiami wszystkich zatwierdzonych i aktualnie stosowanych w jednostce badawczej planów badań i SPO; • weryfikowanie planu badania pod kątem zgodności z zasadami Dobrej Praktyki Laboratoryjnej i za dokumentowanie takiej weryfikacji; • przeprowadzanie inspekcji jednostki badawczej; • przeprowadzenie inspekcji badania oraz inspekcji procesów. • przechowywanie zapisów z każdej inspekcji; • sprawdzenie sprawozdania końcowego w celu potwierdzenia, że stosowane metody, procedury i obserwacje są właściwie i kompletnie opisane, a wyniki podane w sprawozdaniu odpowiadają danym źródłowym uzyskanym w badaniach; • Osoby prowadzące program zapewnienia jakości nie biorą udziału w badaniach, których jakość monitorują!!!

Przyrządy pomiarowe, materiały i odczynniki • są okresowo sprawdzane, czyszczone, konserwowane i wzorcowane, zgodnie z SPO. Czynności te są każdorazowo odnotowane. • Substancje i mieszaniny chemiczne, w tym odczynniki, są oznakowane (tożsamość, okresu ważności i warunków przechowywania).

Systemy badawcze Systemy fizykochemicznych; Systemy biologiczne. • nowo sprowadzone zwierzęta i rośliny poddawane są kwarantannie; • biologiczne systemy badawcze (roślinne i zwierzęce) są aklimatyzowane; • w dniu rozpoczęcia badania systemy biologiczne są wolne od chorób i cech, które mogłyby wpływać na przebieg lub na cele badania; • systemy biologiczne, które podczas badania uległy chorobie lub zostały okaleczone, są izolowane i odpowiednio leczone, są prowadzone zapisy dotyczące pochodzenia, daty otrzymania i stanu biologicznych systemów; • na pomieszczeniach, klatkach lub pojemnikach, w których znajdują się systemy biologiczne, są umieszczane wszystkie informacje niezbędne do identyfikacji tych systemów; • odpowiednie oznakowane, w celu ich identyfikacji; • pomieszczenia, klatki i pojemniki są czyszczone i odkażane z odpowiednią częstotliwością; • systemy badawcze stosowane w badaniach terenowych są rozmieszczone w sposób minimalizujący możliwość wpływu używanych w przeszłości pestycydów lub wpływu zabiegów z użyciem pestycydów z obszarów sąsiadujących.

Materiały badawcze i materiały odniesienia Prowadzi się: • zapisy dotyczące charakterystyki materiałów, dat ich otrzymania i okresu ważności; • ilościową ewidencję materiałów otrzymanych i wykorzystywanych w badaniach. • określenie sposobu postępowania przechowywania i pobierania; • oznaczenie pozwalające na identyfikację materiału, określenie daty ważność i warunków przechowywania; • w każdym badaniu podaje się tożsamość materiału, numer serii, jego skład, czystość oraz stężenie lub inne informacje definiujące każdą serię materiału; • znana jest stabilność materiałów badanych i materiałów odniesienia w stosowanych warunkach i w czasie ich przechowywania oraz badania.

Standardowe procedury operacyjne • udokumentowana procedura określająca sposób przeprowadzenia badania, a także określająca postępowanie nieopisane dokładnie w planach badania lub w wytycznych do badań; • SOP i poprawki do nich są zatwierdzane przez zarządzającego jednostką badawczą • konieczne jest udokumentowane w prowadzonych badaniach odstępstw od SOP • SOP dla: • materiału badanego i materiału odniesienia • przyrządów pomiarowych, • materiałów pomocniczych i odczynników; • w odniesieniu do zapisywania, sporządzania sprawozdań, przechowywania i odzyskiwania danych • w odniesieniu do systemów badawczych systemem badawczym, • w odniesieniu do programu zapewnienia jakości

Przeprowadzenie badania Plan badania – dokument wraz z wszelkimi poprawkami, określający cele i planowany przebieg badania Przed rozpoczęciem każdego badania sporządza się w formie pisemnej plan badania, zweryfikowany pod kątem zgodności z zasadami Dobrej Praktyki Laboratoryjnej przez osobę lub osoby prowadzące program zapewnienia jakości podpisany i opatrzony datą przez kierownika badania i zarządzającego jednostką badawczą. W przypadku badań krótkoterminowych dopuszcza się stosowanie ogólnego planu badania z dołączanym suplementem precyzującym konkretne zadania. Wszelkie zmiany w planie badania podpisuje i opatruje datą kierownik badania. Każde badanie przeprowadzane w jednostce badawczej posiada: odrębny numer, kod lub nazwę identyfikującą to badanie; tym numerem, kodem lub nazwą są oznaczane wszystkie materiały stosowane w tym badaniu.

cd. Przeprowadzenie badania Co zawiera plan badania : • określenie badania, materiału badanego i materiału odniesienia; • daty zatwierdzenia planu badania przez kierownika i zarządzającego jednostką badawczą wraz z ich podpisami; • proponowane daty rozpoczęcia i zakończenia części eksperymentalnej badania • metody badań; • informacje szczegółowe (jeżeli mają zastosowanie); • zapisy – wykaz dokumentów i materiałów, które zostaną zachowane; • informacje dotyczące zleceniodawcy i jednostki badawczej.

cd. Przeprowadzenie badania Badanie : • prowadzone zgodnie z planem badania; • dane uzyskane podczas badania są rejestrowane niezwłocznie i z należytą starannością; • zmiana w danych źródłowych jest dokonywana w sposób umożliwiający odczytanie poprzedniego zapisu; • Stosowane systemy komputerowe zapewniają pełną kontrolę i przechowywanie procesu przetwarzania danych;

Sprawozdania z badań Sprawozdanie końcowe zawiera w szczególności następujące informacje: • identyfikujące badanie, materiał badany i materiał odniesienia • daty – rozpoczęcia i zakończenia części eksperymentalnej badania; • oświadczenie osoby lub osób prowadzących program zapewnienia jakości, • opis materiałów i metod badań • wyniki badań • dotyczące zleceniodawcy i jednostki badawczej. • W sprawozdaniu końcowym zamieszcza się informację o zakresie spełniania zasad Dobrej Praktyki Laboratoryjnej. • Sprawozdanie końcowe podpisuje i opatruje datą kierownik badania. • Korekty sprawozdania końcowego dokonane po jego podpisaniu przez kierownika badania wprowadza się w formie aneksu podpisanego i opatrzonego datą przez kierownika badania. W aneksie wskazuje się wyraźnie powód dokonania korekt. • Z każdego badania sporządza się sprawozdanie końcowe. • Przeredagowanie sprawozdania końcowego w taki sposób, aby było zgodne z wymaganiami jednostek, o których mowa w § 5 ust. 1 pkt 3 rozporządzenia, nie jest dokonaniem korekt lub zmian.

ARCHIWIZACJA Jednostka badawcza przechowuje przez okres 10 lat następujące zapisy i materiały: − plan badania, dane źródłowe, próbki materiału badanego i odniesienia, próbki pobrane z systemów badawczych sprawozdanie końcowe z każdego badania; − raporty wszystkich inspekcji przeprowadzonych przez osobę lub osoby prowadzące program zapewnienia jakości plany działania jednostki; − zapisy dotyczące kwalifikacji, szkolenia i nabytego doświadczenia przez personel badawczy i opis rodzaju wykonywanych przez nich prac; − zapisy i raporty dotyczące konserwacji i wzorcowania wyposażenia pomiarowego; − dokumentację sprawdzania systemów komputerowych; − kopie (NIE!!!) wszystkich edycji standardowych procedur operacyjnych; − dane dotyczące monitorowania środowiska wykonywania badań. Dobra Praktyka Laboratoryjna (DPL) 104

ARCHIWIZACJA § W uzasadnionych przypadkach, zwłaszcza gdy materiał badany lub materiał odniesienia może ulec destabilizacji po okresie trwałości określonym przez producenta lub gdy próby pobrane z systemów badawczych ulegają naturalnemu zniszczeniu i ich stan nie umożliwia ponownej oceny, jest możliwe odstąpienie od wymogu ich przechowywania przez okres 10 lat, z zachowaniem odpowiednio udokumentowanego postępowania. § w przypadku wszystkich badań, oprócz badań krótkoterminowych, są przechowywane próbki każdej partii materiału badanego w celu ich ewentualnej analizy; § Materiał badany i materiał odniesienia są przechowywane w pomieszczeniu archiwum oraz są zewidencjonowane w celu ich łatwego magazynowania i zapewnienia do nich dostępu. Dobra Praktyka Laboratoryjna (DPL) 105

ARCHIWIZACJA § Dostęp do pomieszczenia archiwum mają wyłącznie osoby upoważnione przez zarządzającego jednostką badawczą. § Przychód i rozchód materiału badanego i materiału odniesienia w pomieszczeniu archiwum jest rejestrowany. § Likwidacja jednostki § Przenoszenie archiwum § Kopiowanie danych (nośniki CD) na nowe nośniki § Jednostka, która zostaje usunięta z wykazu certyfikowanych jednostek badawczych zobowiązana jest do przeprowadzenia likwidacji archiwum. Dobra Praktyka Laboratoryjna (DPL) 106

ARCHIWIZACJA Czynnik ryzyka Zapobieganie ryzyka dostęp osób trzecich nieposiadających upoważnienia od zarządzającego jednostką badawczą klucz do archiwum może posiadać jedynie archiwista i zarządzający jednostką badawczą, okno w pomieszczeniu - krata - roleta - folia antywłamaniowa Zalanie/zawilgocenie wietrzenie archiwum w celu uniknięcia kondensacji pary wodnej pożar zabezpieczenia przeciwpożarowe: -czujka dymu -odpowiednia gaśnica (nie niszcząca archiwizowanej dokumentacji) awaria zamrażarki lub lodówki - stosowanie czujników zalania - usunięcie kaloryferów - zabezpieczenie rur - wodoodporne szafy prowadzony monitoring temperatury temperatura powyżej 40°C szkodniki Łódźderatyzacja 20. 10. 2017 r. lub zapobieganie Dobra Praktyka Laboratoryjna (DPL) 107

Krajowy Program Monitorowania 108

KRAJOWY PROGRAM MONITOROWANIA Krajowa Jednostka Monitorująca • W Rzeczypospolitej Polskiej, na mocy przepisów art. 16 ust. 3 ustawy, jednostką właściwą do kontroli i weryfikacji spełniania zasad Dobrej Praktyki Laboratoryjnej przez jednostki badawcze lub certyfikowane jednostki badawcze jest Biuro do spraw Substancji Chemicznych • Kontroli i weryfikacji dokonują inspektorzy Dobrej Praktyki Laboratoryjnej, każdorazowo wyznaczeni przez kierującego Biurem Inspektora do spraw Substancji Chemicznych spośród pracowników Biura, Liczba certyfikowanych jednostek badawczych: • w roku 2004 – 2 • w roku 2019 - 40 (w tym 4 jednostki mające siedzibę poza terytorium Rzeczypospolitej Polskiej: 3 – Chiny; 1 – Chorwacja)

WYKAZ JEDNOSTEK BADAWCZYCH, KTÓRE UZYSKAŁY CERTYFIKATY ZGODNOŚCI Z ZASADAMI DOBREJ PRAKTYKI LABORATORYJNEJ OD POLSKIEJ JEDNOSTKI DS. MONITOROWANIA DPL – Biura do spraw Substancji Chemicznych

KRAJOWY PROGRAM MONITOROWANIA • Certyfikowane jednostki badawcze podlegają okresowej lub doraźnej kontroli i weryfikacji spełniania zasad DPL • Kontrola i weryfikacja mogą również następować na wniosek jednostek właściwych do spraw kontroli i weryfikacji spełniania zasad Dobrej Praktyki Laboratoryjnej w państwach, o których mowa w ust. 6, lub jednostek właściwych do spraw Dobrej Praktyki Laboratoryjnej w Komisji Europejskiej i w OECD • W przypadku niespełniania zasad DPL przez certyfikowaną jednostkę badawczą Inspektor, w drodze decyzji, cofa wydany certyfikat i wykreśla jednostkę z wykazu certyfikowanych jednostek badawczych • W przypadku stwierdzenia, że określone badanie lub badania zostały wykonane niezgodnie z zasadami Dobrej Praktyki Laboratoryjnej, Inspektor, w drodze decyzji, stwierdza niespełnianie zasad DPL w odniesieniu do określonego badania lub badań Krajowy Program Monitorowania 111

KRAJOWY PROGRAM MONITOROWANIA • Kontrolę i weryfikację spełniania zasad Dobrej Praktyki Laboratoryjnej, przeprowadza się na podstawie pisemnego upoważnienia wydanego przez Inspektora do spraw Substancji Chemicznych, które zawiera: ‒ nazwisko i imię oraz numer dokumentu potwierdzającego tożsamość inspektora Dobrej Praktyki Laboratoryjnej lub osoby, o której mowa w ust. 3, dokonującej kontroli i weryfikacji; ‒ nazwę jednostki badawczej albo certyfikowanej jednostki badawczej, w której jest dokonywana kontrola i weryfikacja; ‒ datę przeprowadzenia kontroli i weryfikacji, określenie ich zakresu i przewidywanego czasu trwania. Krajowy Program Monitorowania 112

KRAJOWY PROGRAM MONITOROWANIA • Inspektorzy DPL oraz osoby, dokonujące kontroli i weryfikacji są uprawnieni do: ‒ wstępu na teren nieruchomości, obiektów i lokali jednostki badawczej albo certyfikowanej jednostki badawczej, w której jest dokonywana kontrola i weryfikacja, w dniach i godzinach jej pracy, ‒ wglądu do dokumentacji, w tym do danych źródłowych, oraz żądania informacji i wyjaśnień dotyczących wykonywanych przez jednostkę badawczą albo certyfikowaną jednostkę badawczą badań substancji lub ich mieszanin, ‒ czynności kontroli i weryfikacji są dokonywane w obecności upoważnionego przedstawiciela jednostki badawczej albo certyfikowanej jednostki badawczej, w której jest dokonywana kontrola i weryfikacja. Krajowy Program Monitorowania 113

KRAJOWY PROGRAM MONITOROWANIA W przypadku gdy w trakcie kontroli i weryfikacji inspektorzy Dobrej Praktyki Laboratoryjnej lub inne osoby wyznaczone przez Inspektora do przeprowadzenia kontroli i weryfikacji mają dostęp do informacji stanowiących tajemnicę przedsiębiorstwa w rozumieniu art. 11 ust. 4 ustawy z dnia 16 kwietnia 1993 r. o zwalczaniu nieuczciwej konkurencji (Dz. U. z 2003 r. Nr 153, poz. 1503, z późn. zm. 1), informacje takie nie mogą zostać ujawnione. W przypadku gdy takie informacje są zawarte w protokole z przeprowadzonej kontroli i weryfikacji, protokół może zostać ujawniony na wniosek odpowiednich władz krajowych, Komisji Europejskiej, jednostek badawczych lub certyfikowanych jednostek badawczych, w których przeprowadzono kontrolę i weryfikację, oraz gdy dotyczy to określonego badania – zlecającego to badanie. Krajowy Program Monitorowania 114

KRAJOWY PROGRAM MONITOROWANIA • Kontrola i weryfikacja spełniania zasad Dobrej Praktyki Laboratoryjnej przez jednostki badawcze w przypadku, o którym mowa w: ‒ podlegają opłacie jednorazowej, której dowód uiszczenia jednostka badawcza przedkłada wraz z wnioskiem o wydanie certyfikatu i wpis do wykazu certyfikowanych jednostek badawczych, ‒ podlegają stałej opłacie rocznej, ‒ w przypadku uzyskania certyfikatu i wpisu do wykazu certyfikowanych jednostek badawczych opłata jednorazowa, o której mowa w ust. 1 pkt 1, staje się opłatą roczną, o której mowa w ust. 1 pkt 2, za rok, w którym jednostka badawcza uzyskała certyfikat, ‒ w przypadku nieuiszczenia stałej opłaty rocznej, o której mowa w ust. 1 pkt 2, Inspektor, w drodze decyzji, cofa wydany certyfikat i wykreśla certyfikowaną jednostkę badawczą z wykazu, o którym mowa w art. 16 ust. 4. Krajowy Program Monitorowania 115

KRAJOWY PROGRAM MONITOROWANIA • Aktualny wykaz certyfikowanych jednostek badawczych oraz krajowy program monitorowania zgodności z zasadami znajduje się w BIP Biura do spraw Substancji Chemicznych https: //www. chemikalia. gov. pl/czym_jest_dobra_praktyka_laboratoryjna. html • W przypadku jednostki badawczej mającej siedzibę poza terytorium Rzeczypospolitej Polskiej uznaje się, że jednostka ta spełnia zasady Dobrej Praktyki Laboratoryjnej, po przedstawieniu ważnego certyfikatu lub innego właściwego dokumentu nadanego tej jednostce przez jednostkę właściwą do kontroli i weryfikacji spełniania zasad Dobrej Praktyki Laboratoryjnej w państwach OECD lub w innych państwach, w których ustanowiono w porozumieniu z OECD takie jednostki. Krajowy Program Monitorowania 116

KRAJOWY PROGRAM MONITOROWANIA KONTAKT PROŚBA DO INSPEKTORA ds. SCH ? ? ? KONTROLA PRZEDWSTĘPNA ? ? ? UWAGI ? ? ? PROTOKÓŁ Z KONTROLI WSTĘPNEJ 14 D KONTROLA WSTĘPNA 7 D PISMO INFORMUJĄCE O WST. KONTROLI I WERYFIKACJI 14 D ? ? ? WNIOSEK DO INSPEKTORA DS. SCH 30 D DECYZJA 14 D KONTROLA I WERYFIKACJA JEDNOSTKI BADAWCZEJ 14 D PROTOKÓŁ Z KONTROLI I WERYFIKACJI 2 LATA Krajowy Program Monitorowania ? ? ? WNIOSEK JEDNOSTKI BADAWCZEJ

KRAJOWY PROGRAM MONITOROWANIA USTALENIE TRMINU KONTROLI I WERYFIKACJI PISMO INFORMUJĄCE O KONTROLI I WERYFIKACJI ? ? ? 7 D KONTROLA I WERYFIKACJA 14 D ? ? ? DECYZJA/ CERTYFIKAT 14 D 30 D WNIOSEK DO INSPEKTORA DS. SCH 2 LATA ODWOŁANIE DO INSPEKTORA DS. SCH 14 D KONTROLA I WERYFIKACJA MINISTER ZDROWIA 30 D 14 D DODATKOWE ZAPYTANIA DO JEDNOSTKI ? ? ? 14 D ZASTRZEŻENIA DO PROTOKOŁU ODPOWIEDŹ JEDNOSTKI 14 D PROTOKÓŁ/ LISTA DOKUMENTÓW STANOWISKO INSPEKTORA ds. SCH ? ? ? 14 D PODTRZYMANIE DECYZJI 14 D ZMIANA DECYZJI Krajowy Program Monitorowania 118

KONTROLA I WERYFIKACJA 119

KONTROLA I WERYFIKACJA SPEŁNIANIA PRZEZ JEDNOSTKĘ ZASAD DPL • Kontrolę jednostki badawczej lub certyfikowanej jednostki badawczej przeprowadza się w siedzibie jednostki lub w miejscu przeprowadzania badania, a weryfikację można przeprowadzić również poza tymi miejscami. • W ramach kontroli i weryfikacji: – sprawdza się strukturę zarządzania oraz procedury operacyjne; – przeprowadza się wywiady z personelem; – ocenia się integralność danych otrzymanych w jednostce badawczej; – ocenia się stopień zgodności z zasadami Dobrej Praktyki Laboratoryjnej. Rewizję badań przeprowadza się przez porównanie danych źródłowych i towarzyszących zapisów ze sprawozdaniem końcowym lub okresowym. 120

KONTROLA I WERYFIKACJA SPEŁNIANIA PRZEZ JEDNOSTKĘ ZASAD DPL Rodzaje kontroli • Wstępna • Okresowa kontrola i weryfikacja • Zarządzana doraźnie przez Inspektora do spraw Substancji Chemicznych z własnej inicjatywy lub na wniosek jednostek właściwych do spraw kontroli i weryfikacji spełniania zasad DPL w państwach członkowskich Unii Europejskiej (ECHA, EMA, EFSA), państwach OECD • Kontrola sprawdzająca, czy odstępstwa od spełniania zasad DPL zostały usunięte 121

Jak sprawdzić czy badanie jest wykonane w zgodności z DPL? Czy jednostka znajduje się w wykazie jednostek certyfikowanych? Jaki zakres posiada jednostka? Czy do sprawozdania załączono oświadczenie KB? Czy do sprawozdania załączono oświadczenie JZJ?

Dziękuję za uwagę
- Slides: 123