Yahya Ahmed Yahya Hemostasis Cascade Model of Coagulation

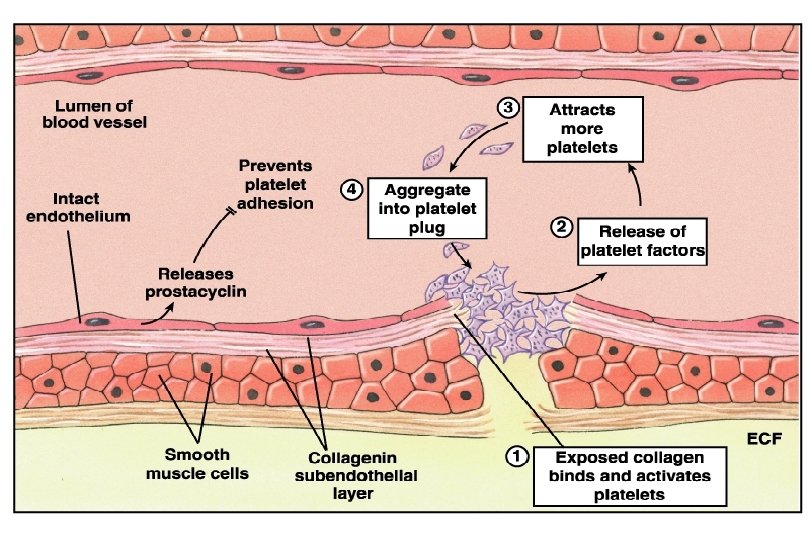
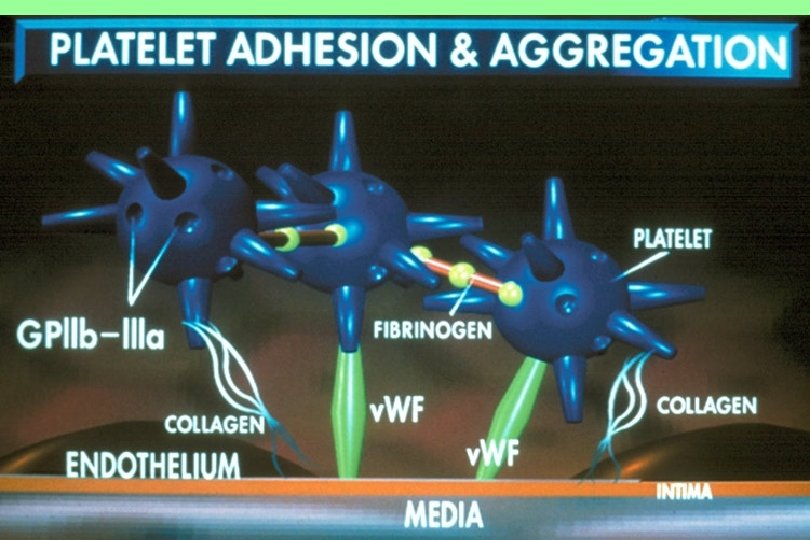


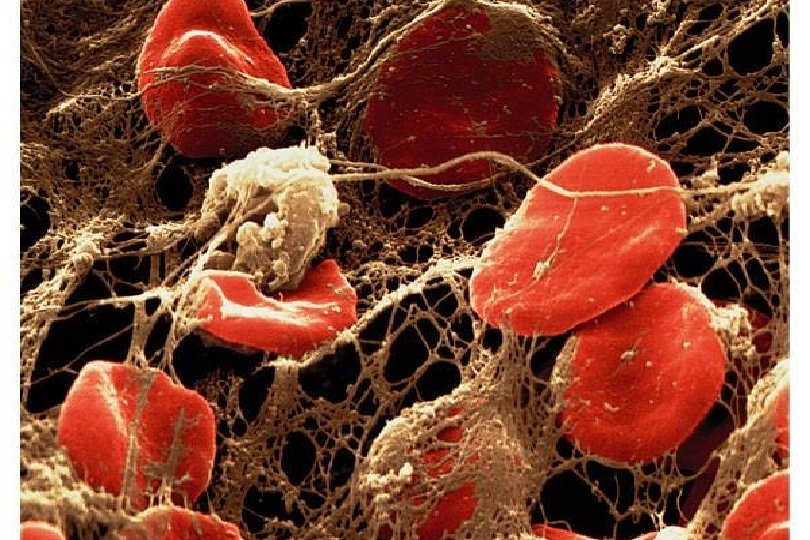


 n 1. 2. Platelet Disorders Thrombocytopenia Platelet function disorders (cong.")

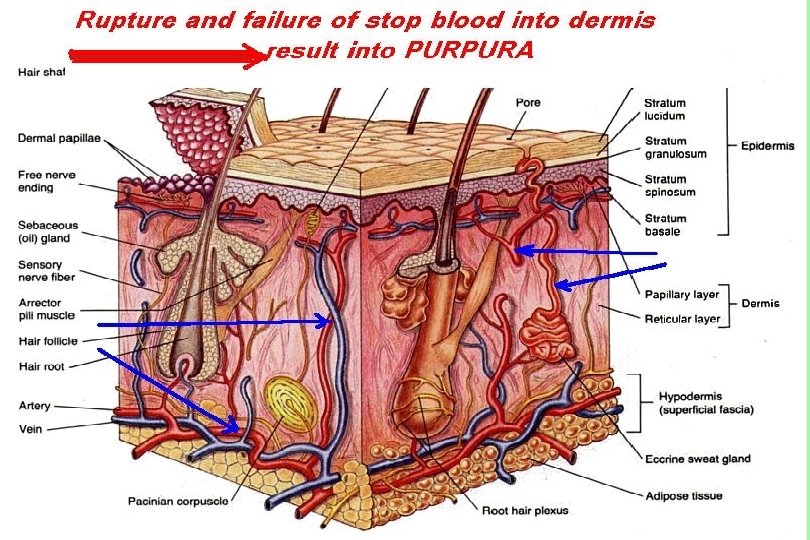
")


 occur mainly in")
 elevated n Prothrombin time (PT) normal n Decreased factor")


: • Type 1, partial quantitative deficiency (≈70%) •")







. n Hypofibrogenrmia")




 Splenomegaly Abnormal BM combined Splenomegaly normal BM abnormal sequestration No")
 n n n n The presence of excess antibodies against")





")
 Senile Purpura A disorder affecting older patients, particularly those who")
 Hereditary Hemorrhagic Telangiectasia It is an autosomal dominant disorder with")
 n Hemorrhage may be a prominent feature of scurvy (Vitamin")




- Slides: 61
Yahya Ahmed Yahya
Hemostasis
Cascade Model of Coagulation
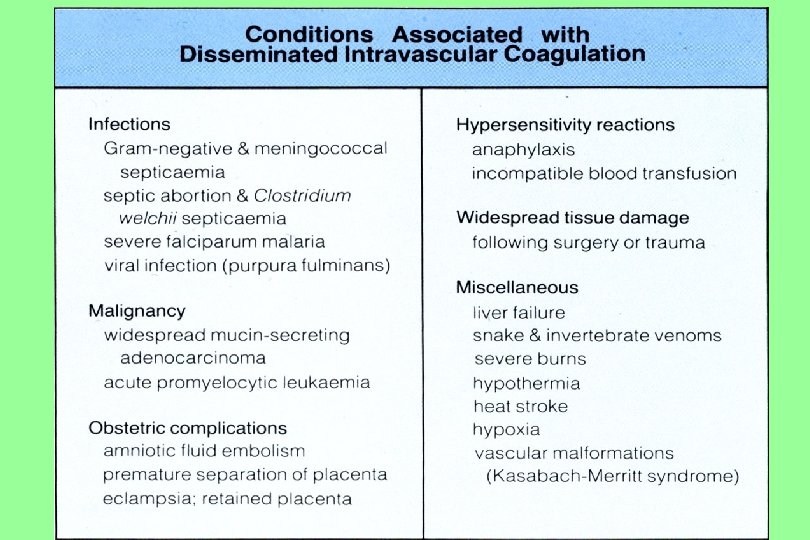
Bleeding Disorders n 1. 2. Congenital Clotting Disorders Hemophilia A and B Von Willibrand’s Disease Acquired Clotting Disorders Disseminated Intravascular Coagulation Liver Disease
Disorders of Hemostasis n Vascular disorders – • Senile purpura, easy bruising, Henoch-Schonlein purpura. n Platelet disorders • Quantitative - Thrombocytopenia • Qualitative - Platelet function disorders – Glanzmans n Coagulation disorders • Congenital - Haemophilia (A, B), Von-Willebrands • Acquired - Vitamin-K deficiency, DIC, Liver disease n Mixed/Consumption: DIC
Bleeding Disorders (contin. ) n 1. 2. Platelet Disorders Thrombocytopenia Platelet function disorders (cong. & acquir. ) Vascular wall Disorders Henoch’s Schoenlein Purpura Senile Purpura
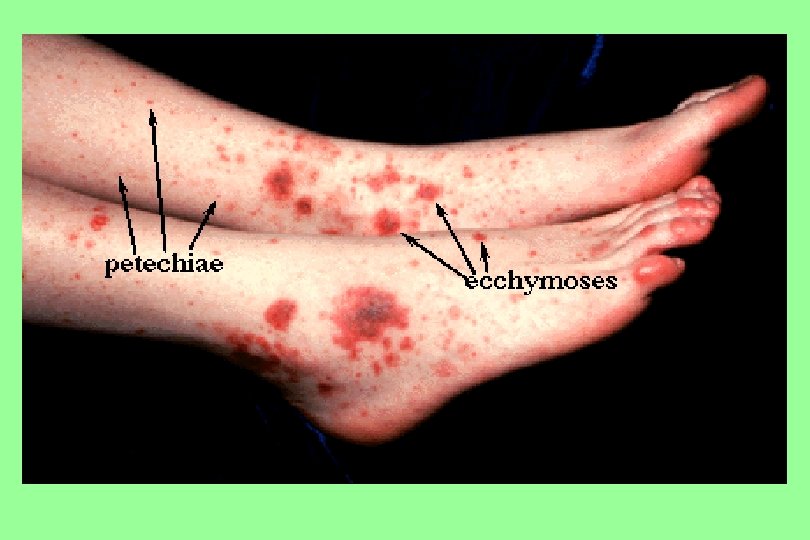
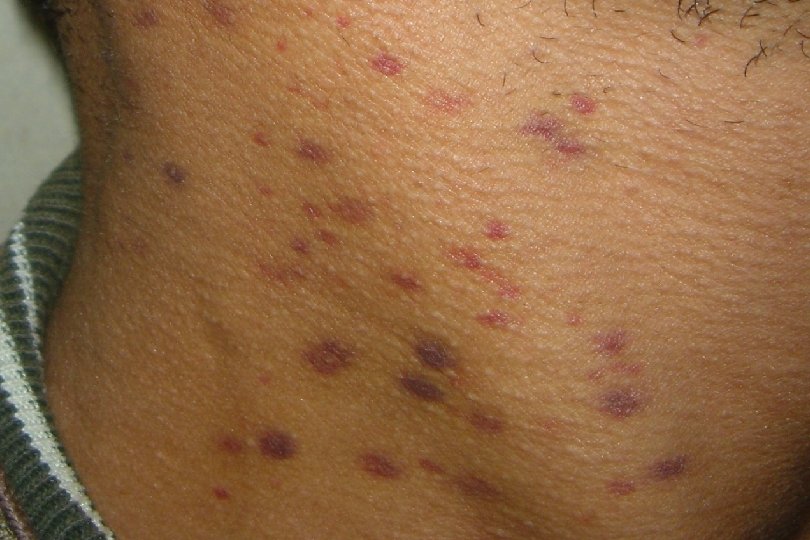
Definitions n n Petechiae are non-raised, round, purplish-red dots caused by intradermal hemorrhage. . They are pinpoint hemorrhages seen in the skin subsequent to capillary or arteriole rupture. Purpura (purple or brown-red) are larger often less regular areas of bleeding in the skin. Ecchymoses are even larger, non-regular in shape, non-elevated areas commonly called bruises. In the skin, the bruise changes color from purple, to blue, to green to yellow as the free hemoglobin is metabolized and changes its oxidation levels. Hematomas are small to large volumes of blood trapped in soft tissues or tissue spaces; they often are in locations where they compress tubular organs. They are due to blood vessel rupture.
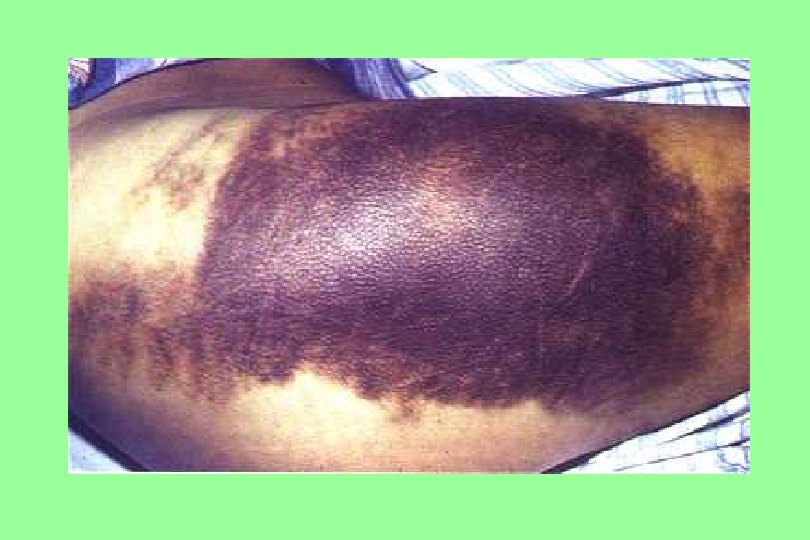
Ecchymoses (typical of coagulation factor disorders)
Congenital Bleeding Disorders
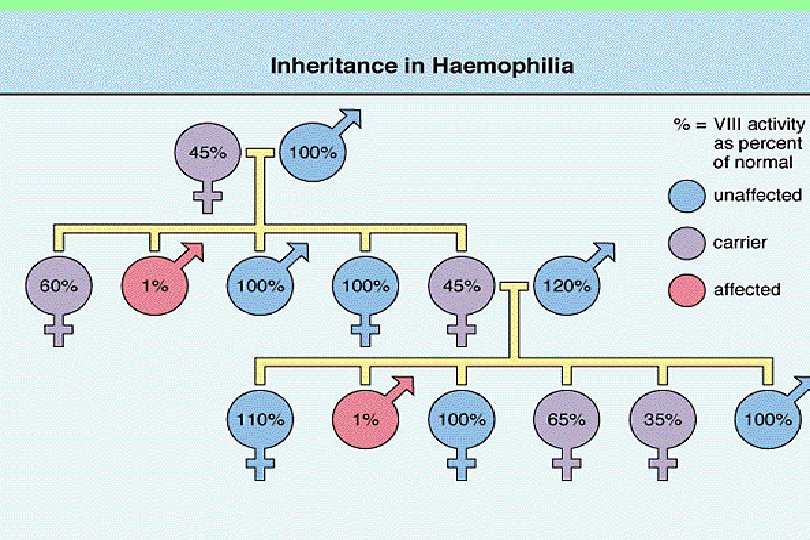
Hemophilia A and B Coagulation factor deficiency Inheritance Incidence Severity Complications Hemophilia A Hemophilia B Factor VIII Factor IX X-linked recessive 1/10, 000 males 1/50, 000 males Related to factor level <1% - Severe - spontaneous bleeding 1 -5% - Moderate - bleeding with mild injury 5 -25% - Mild - bleeding with surgery or trauma Soft tissue bleeding
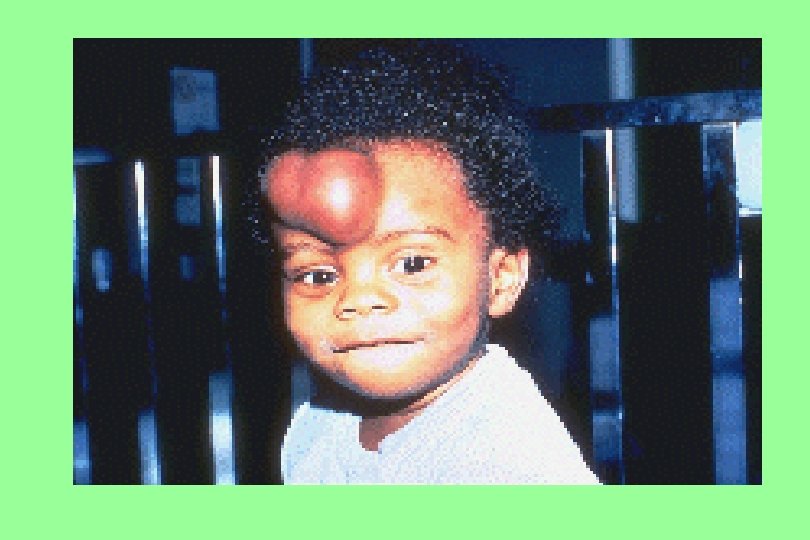
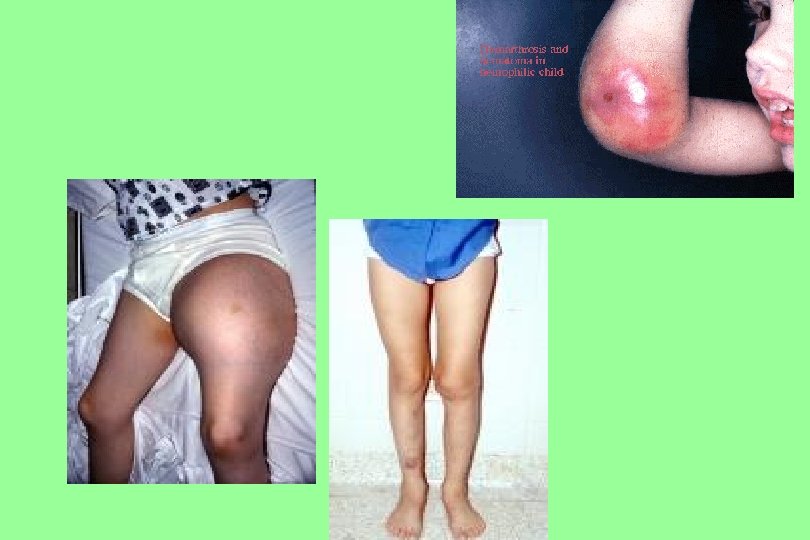
Hemophilia A The most common hereditary coagulation disorder (1: 10, 000) occur mainly in men. Women may be carriers, they have below-normal levels of Factor VIII in their blood. Symptoms • Excessive bleeding from slight trauma, Dental extraction, Surgery • Visceral bleeding • Hemarthrosis • Contractures • Degenerative arthritis Other sites of bleeding Urinary tract CNS, neck (may be life-threatening) Prolonged bleeding after surgery or dental extractions
DIAGNOSIS Partial Thromboplastin time (PTT) elevated n Prothrombin time (PT) normal n Decreased factor VIII level n
Haemarthrosis
Von Willebrand's Disease Von Willebrand factor mediates platelet adhesion and act as a carrier for factor VIII. n It is the most common inherited bleeding disorder. It is autosomal dominant in most patients. n Mild bleeding disorder (often undiagnosed). n Symptoms and Signs Severe Menorrhagia (common presentation in women). It is commonly 1 st discovered several days after delivery. Lab. : bleeding time, factor 8 level, pl. agg. to restocetine. Treatment: Factor 8 concentrate or fresh frozen plasma.
Von Willebrand Disease (3 major types): • Type 1, partial quantitative deficiency (≈70%) • Type 2, qualitative abnormalities (≈ 25%) • Type 3, complete deficiency (< 5%)
Acquired Clotting Disorders
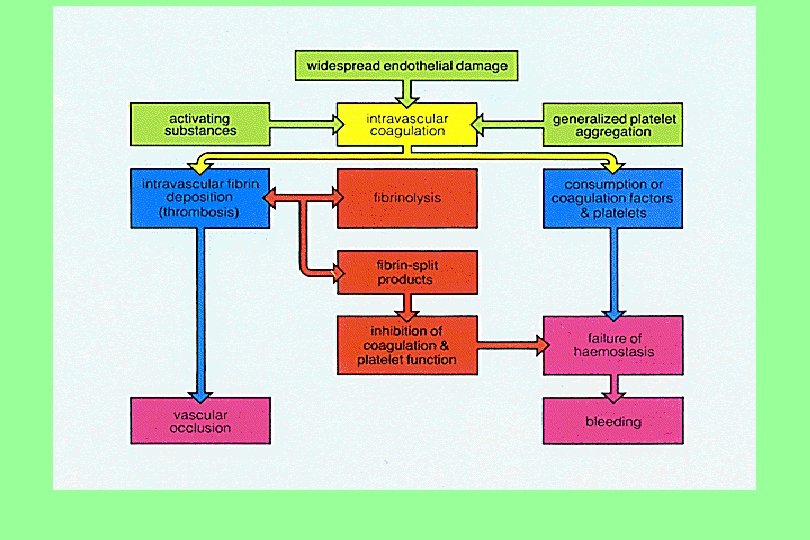
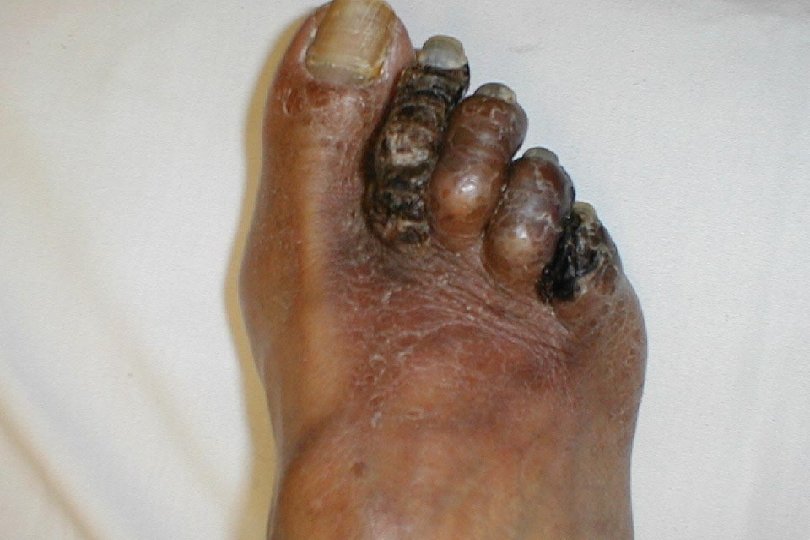
Clinical picture of DIC Pathophysiology n Acute: Consumptive coagulopathy. n Chronic: Micro emboli or thrombi in vasculature. Signs and symptoms Profuse bleeding from many sites. n Multi-organ Failure (thromboembolic). n
Pathogenesis of DIC
The diagnosis of DIC : n Prolonged clotting and prothrombin times. n Decreased levels of individual clotting factors, fibrinogen and platelet count. n Increased fibrin degradation products (d. Dimer).
TREATMENT of DIC n The primary treatment of disseminated intravascular coagulation is successful therapy for the underlying condition (shock, sepsis, cancer, . . ). n Replacement of the missing coagulation factors is often necessary by infusion of fresh frozen plasma and platelets.
Bleeding tendency in liver diseases n Hypoprothrmbinemia (HC failure and/or obstructive jaundice). n Hypofibrogenrmia (chronic DIC). n Activated fibrinolysis due to less inactivation by diseased liver. n Thrombocytopenia due to chronic DIC and/or hypersplenism.
Defect in Platelet Function Thrombathenia Number Thrombocytopenia Congenital : VW disease Bernard Soulier synd. Glansman's thrombasthenia Vascular Defect Acquired NSAID as ibubrufen, aspirin, . . Myelo-proliferative disorders Multiple myeloma Renal failure
n Normal Count range from 150, 000 to 400, 000 per cu/ml. n Mild Thrombocytopenia >150000 to 50000. n Moderate Thrombocytopenia > 50000 to 10000. n Severe Thrombocytopenia > 10000.
Causes of Thrombocytopenia Decreased production: 1. 2. 3. Aplastic anemia Bone marrow infilteration by MALIGNANCY OR FIBROSIS Rare congenital disorders: Wiskott Aldrich - tiny platelets Increased destruction or consumption: 1. 2. 3. Idiopathic: ITP. DIC &TTP&HUS. Valve prothesis, haemodialysis and open cardiac surgery. Redistribution: Hypersplenism. Dilutional: Over-infusion of platelet-free fluids as crystaloids, pooled plasma or banked blood.
Causes of Thrombocytopenia (cont) Splenomegaly Abnormal BM combined Splenomegaly normal BM abnormal sequestration No splenomegaly Abnormal BM production defect No splenomegaly normal BM excess destruction disorder Leukemias Lymphomas Hypersplenism Liver disease Congestive splenomegaly Acute DIC (falsiparum malaria) Myloid metaplasia storage diseases ITP Dilutional Aplastic or megaloplastic BM Drug-induced immune thrombocytopenia myelodysplastic syndrome Post-transfusion purpura Metastatic cancer TTP & HUS
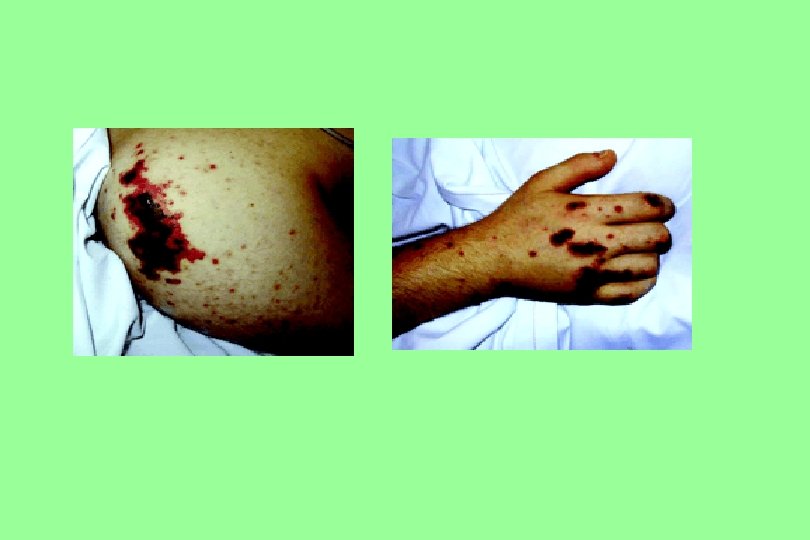
Idiopathic Thrombocytopenic Purpura (ITP) n n n n The presence of excess antibodies against platelets that break them down at an abnormal rate. ITP may be accompanied by autoimmune haemolytic anaemia (Evans’ syndrome). Bleeding usually appears as pinpoint bleeding spots (petechiae) in the skin and mucous membranes. The disease occur in two forms Acute that occurs after infections in children and newborn infants. Chronic ITP occurs most commonly in females in the age range 20 – 40 years. Mild Splenomegaly in 5 to 10% of cases Absent signs: No fever, lethargy, pallor or weight loss, No bone or joint pain. No Lymphadenopathy and No hepatomegaly. DIAGNOSIS : besides thrombocytopenia bone marrow examination show deficient megakaryocytes with deficient platelet budding.
TREATMENT of ITP The goal of medical care is to increase the platelet count to a safe level (greater than 30, 000/mm ) 1. Adrenal steroids in chronic ITP 2. IVIG (immune gamma globulin) or intravenous Anti-D immune globulin used to 3. 4. safe acute ITP or before op. in chronic severe cases. Splenectomy (after 3 to 6 months ) is necessary in some cases if the patient continues to require 10 to 20 mg/day of prednisone to keep the platelet count greater than 30, 000/mm or when other treatments failed. Platelet transfusions are used under cover of steroids when thrombocytopenia is dangerous for patients' life
HYPERSPLENISM Definition n It is a type of disorder which causes the spleen to rapidly and prematurely destroy blood cells. n An enlarged spleen can be caused by a variety of diseases. Diagnosis Blood tests indicate decreases in white blood cells, red blood cells, and/or platelets. Injection of a radioactive Cromium indicates areas where the spleen is holding on to large numbers of red cells or is destroying them.
Vascular Purpura • Vascular disorders may cause petechiae, purpura, and bruising but seldom lead to serious blood loss. • Henoch's schoenline Purpura (post infective type III hypersensitivity) • Senile purpura • Ehler Danlos syndrome • Factetios(self-inflected) • Autoerythrocyte sensitization • Scurvy (severe vit. C deficiency)
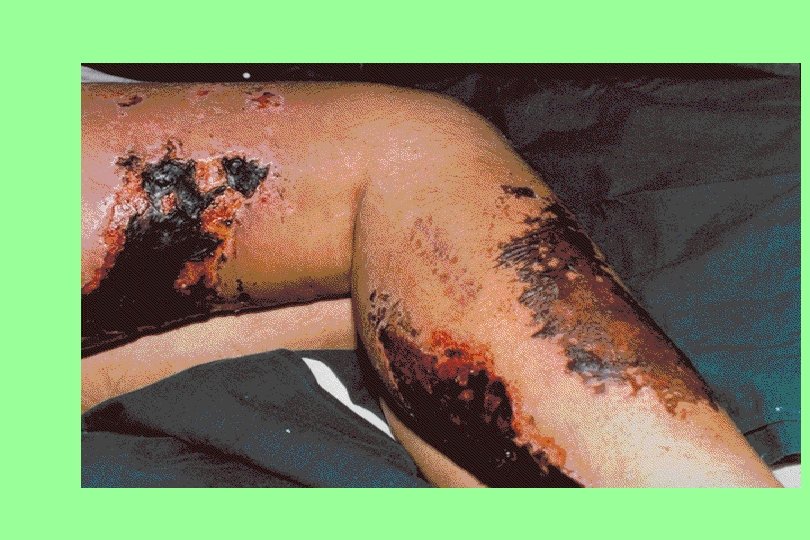
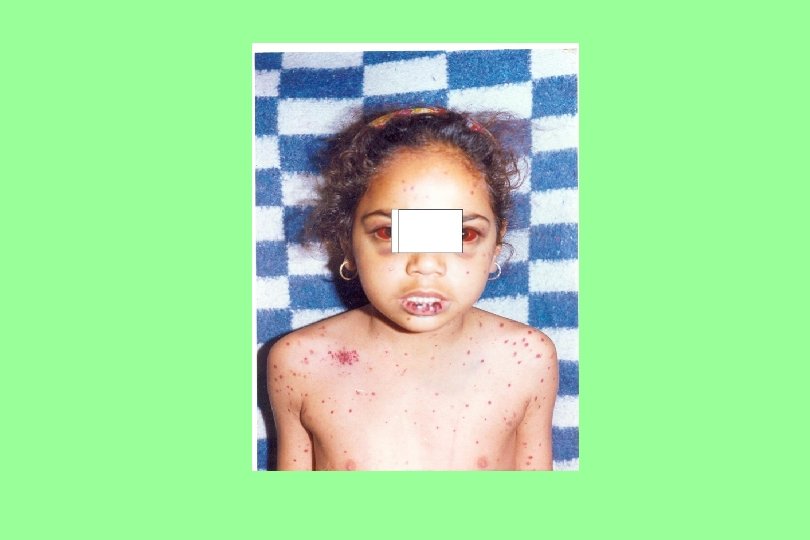
Henoch-Schonlein purpura 20 y Male, fever, painful symmetric polyarthritis for a day. During the next two days, edema and palpable purpura developed.
Vascular Purpura (cont. )
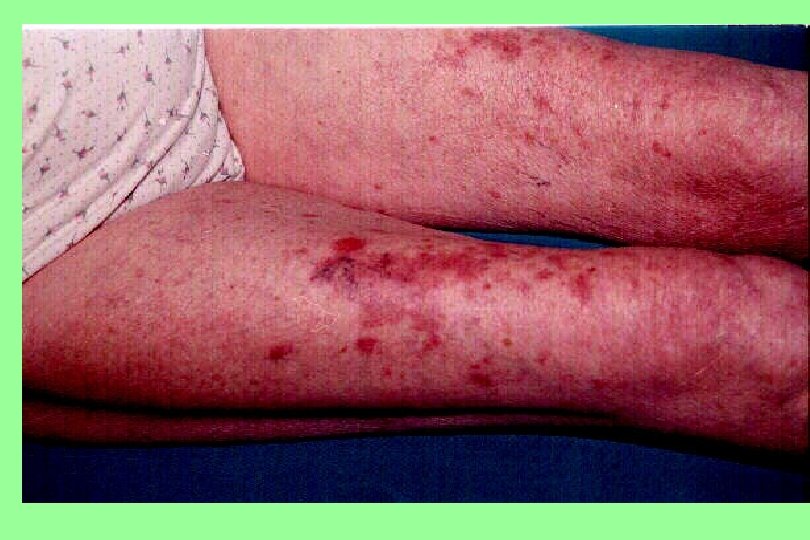
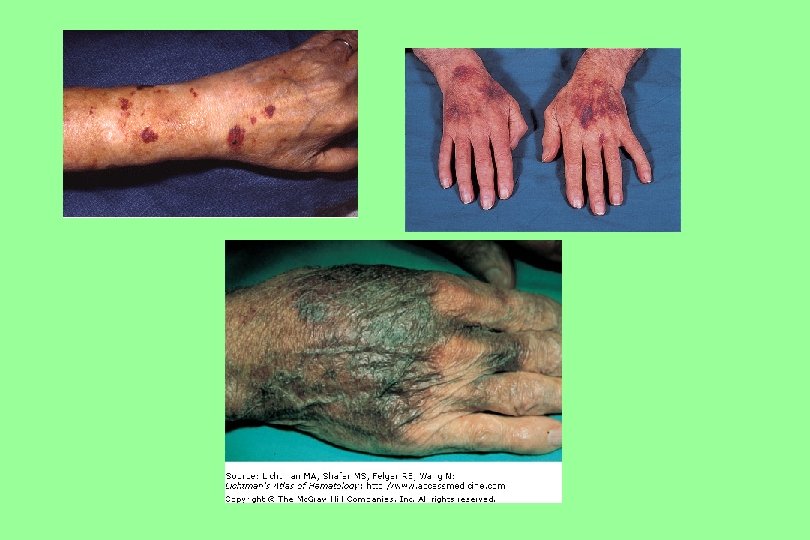
Vascular Purpura (cont. ) Senile Purpura A disorder affecting older patients, particularly those who have had excessive sun exposure, in whom dark purple ecchymoses. Characteristically confined to the extensor surfaces of the hands and forearms, persist for a long time.
Vascular Purpura (cont. ) Hereditary Hemorrhagic Telangiectasia It is an autosomal dominant disorder with variable penetrance characterized by epistaxis, mucocutaneous telangiectases, and visceral arteriovenous malformations. Characteristically confined to the extensor surfaces of the hands and forearms, persist for a long time.
Vascular Purpura (cont. ) n Hemorrhage may be a prominent feature of scurvy (Vitamin C Deficiency). n In vascular bleeding disorders, tests of hemostasis are usually normal. The diagnosis is made from other clinical findings.
Laboratory Approach to Coagulation Disorders
ANY QUESTIONS?
THANK YOU