WRODZONE WADY METABOLIZMU what we learn from rare

WRODZONE WADY METABOLIZMU „…what we learn from rare disorders often has profound consequences for our understanding of more common conditions. ” Francis S. Collins Director of the National Human Genome Research Institute
,")
DEFINICJA • zaburzenia powstałe w wyniku genetycznie uwarunkowanych bloków enzymatycznych (mutacje w pojedynczym genie), prowadzące do nieprawidłowych procesów przemiany materii • ponad 3000 wrodzonych wad metabolizmu

CZĘSTOŚĆ WYSTĘPOWANIA • fenyloketonuria: 1: 8000; galaktozemia: 1: 40, 000. • choroba syropu klonowego 1: 250, 000 homocystynuria 1: 250, 000 • inne 1: 1, 000
 • W Polsce rodzi się 150")
Wrodzone wady metabolizmu Inborn Errors of Metabolism (IEM) • W Polsce rodzi się 150 -350 nowych przypadków rocznie, z czego ok. 40 -50 jest rozpoznawanych • Najczęściej AR, pokrewieństwo rodziców ? • Pierwsze objawy kliniczne u noworodka w 2/3 przypadków IEM

SPOSÓB DZIEDZICZENIA • AR • AD • sprzężone z X • dziedziczenie mitochondrialne

TYPY WRODZONYCH WAD METABOLIZMU - I • Metabolizm aminokwasów fenyloketonuria, alkaptonuria, homocystynuria, choroba syropu klonowego • Defekty cyklu mocznikowego • Metabolizm wodorowęglanów metabolizm monosacharydów choroby spichrzeniowe glikogenu • Metabolizm steroidów wrodzony przerost nadnerczy, brak wrażliwości na androgeny • Metabolizm lipidów rodzinna hipercholesterolemia, • Mukopolisacharoidozy zespół Hurlera, zespół Huntera, zespół Sanfilipo, zespół Morquio • Sfingolipidozy choroba Tay-Sachsa, choroba Gaucher, choroba Niemann-Picka choroba Wilsona, choroba Menkesa

TYPY WRODZONYCH WAD METABOLIZMU - II • Metabolizm puryn i pirymidyn choroba Lesch-Nyhana • Metabolizm porfiryn porfirie wątrobowe, erytropoetyczne • Zaburzenia dot kwasów organicznych kwasica metylomalonowa, kwasica propionowa • Metabolizm miedzi choroba Wilsona, choroba Menkesa • Choroby peroksyzomalne zespół Zellwegera, adrenoleukodystrofia • Choroby mitochondrialne • Zaburzenia oksydacji kwasów tłuszczowych acyduria glutarowa

OBJAWY KLINICZNE U chorego noworodka zawsze należy podejrzewać wadę metabolizmu

Tabele na podstawie: Archives of disease in childhood fetal and neonatal edition. Chakrapani et al. . , 2001; 84; F 205 -F 210

OBJAWY KLINICZNE • Zaburzenia neurologiczne - encefalopatia i drgawki *po urodzeniu zdrowe, w krótkim czasie senność, gorsze łaknienie, wymioty, niepokój, często wysokie stężenie NH 3, zasadowica oddechowa, ketonuria acydurie organiczne: propionowa, metylomalonowa, izowalerianowa; zaburzenia cyklu mocznikowego *po urodzeniu utrata przytomności, drgawki, bezdech nieketotyczna hiperglicynemia, drgawki pirydoksynowe, niedobór kofaktora molibdenowego, pierwotna kwasica mleczanowa; choroby mitochondrialne i peroksyzomalne


OBJAWY KLINICZNE • Kwasica metaboliczna przedłużająca się kwasica o niewyjaśnionej przyczynie zwiększona wartość luki anionowej acydemie organiczne • Kwasica mleczanowa zaburzenia metabolizmu pirogronianów, defekty łańcucha oddechowego zaburzenia oksydacji kwasów tłuszczowych, kwasice organiczne, zaburzenia cyklu mocznikowego Stężenie mleczanów powyżej 3 mmol/l bez zamartwicy, uszkodzeń narządów - diagnostyka w kierunku wrodzonych wad metabolizmu

OBJAWY KLINICZNE • Hipoglikemia pobrać materiał do badań w trakcie epizodu hipoglikemii zaburzenia oksydacji tłuszczów, wąrobowe postacie glikogenoz, zaburzenia glukoneogenezy Wtórna hipoglikemia w chorobach metabolicznych pierwotnie zaburzających funkcje wątroby *po urodzeniu utrata przytomności, drgawki, bezdech nieketotyczna hiperglicynemia, drgawki pirydoksynowe, niedobór kofaktora molibdenowego, pierwotna kwasica mleczanowa; choroby mitochondrialne i peroksyzomalne


OBJAWY KLINICZNE • Zaburzenia czynności wątroby najczęstsza przyczyna metaboliczna - galaktozemia tyrozynemia wątrobowo-nerkowa niedobór alpha 1 – antytrypsyny hemochromatoza noworodków mitochondrialne zaburzenia łańcucha oddechowego typ C choroby Niemanna i Picka


OBJAWY KLINICZNE • Objawy ze strony serca *niewydolność serca, kardiomiopatia przerostowa, hipotonia mitochondrialne zaburzenia łańcucha oddechowego zaburzenia oksydacji długołańcuchowych kwasów tłuszczowych, choroba Pompego (glikogenoza typ II) *kardiomiopatia i/lub płyn w osierdziu + zaburzenia rozwoju fizycznego, dysmorfia twarzy, wciągnięcie brodawek sutkowych, nieprawidłowe rozmieszczenie tkanki tłuszczowej wieloukładowe wrodzone zaburzenia glikozylacji (CDG) *kardiomiopatia rozstrzeniowa, neutropenia, zaburzenie stężenia kwasów organicznych w moczu (zespół Bartha)
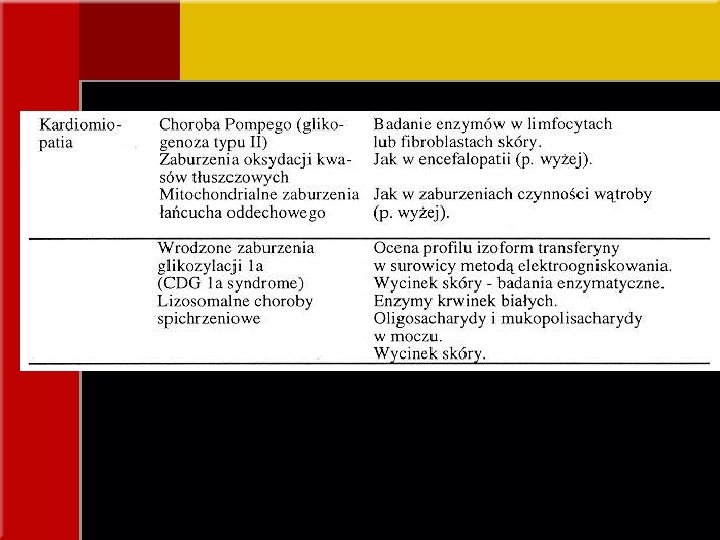

OBJAWY KLINICZNE • Dysmorfia lizosomalne choroby spichrzeniowe - pogrubinie rysów twarzy zaburzenia syntezy steroli (SLOS) wrodzone zaburzenia glikozylacji acyduria glutarowa typu II, acyduria 3 hydroksyizomasłowa, tj. wpływające bezpośrednio na metabolizm energetyczny (wady serca, nerek, układu kostno-szkieletowego)

OBJAWY KLINICZNE • Inne choroba syropu klonowego zapach spoconych stóp - kwasica izowalerianowa, acyduria glutarowa typu II niestabilność temperatury ciała – wczesny objaw zespółu Menkesa uogólniony nieimmunologiczny obrzęk płodu – wiele chorób metabolicznych żółtaczka, skaza krwotoczna – uszkodzenia wątroby, zaburzenia cyklu mocznikowego

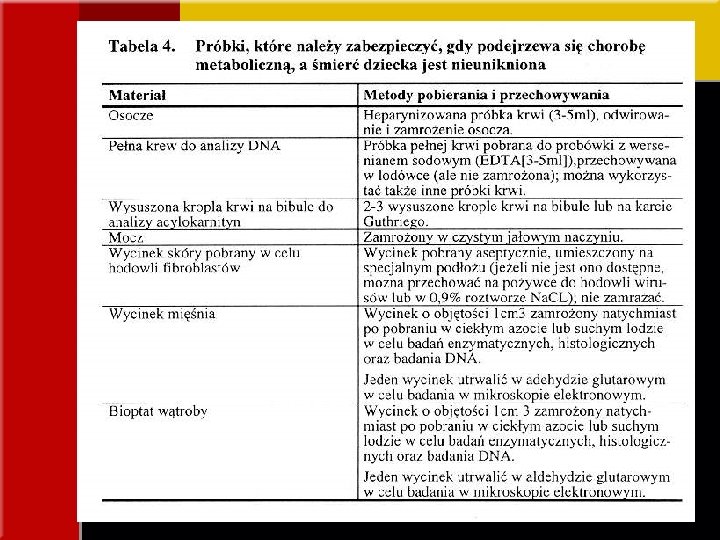

BADANIA PRZESIEWOWE

Metabolizm aminokwasów

Fenyloketonuria mutacja w genie kodującym hydroksylazę fenyloalaniny PAH toxic

Fenyloketonuria Pierwsza wrodzona wada metabolizmu rutynowo diagnozowana – podwyższone stężenie fenyloalaniny – podwyższone stężenie kw. fenylopirogronowego w moczu Opóźnienie rozwoju psychoruchowego - pierwszy objaw sugerujący chorobę. Z wiekiem dziecka opóźnienie rozwoju umysłowego zwykle w stopniu głębokim (iloraz inteligencji 20 -40).
 we krwi => uszkodzenie OUN * zmiany skórne")
OBJAWY FENYLOKETONURII * wzrost fenyloalaniny (Phe) we krwi => uszkodzenie OUN * zmiany skórne (o charakterze zmian alergicznych lub zapalnych) * skłonność do wymiotów (sugerująca zwężenie odźwiernika) * wzmożona pobudliwość, spadek napięcia mięśniowego * wzmożenie odruchów ścięgnistych * drgawki (głównie pod postacią napadów zgieciowych) * zbyt wolne przyrosty obwodu głowy - małogłowe (60 -90% chorych)! * opóźnienie rozwoju psychoruchowego * „rozcieńczenie barwnika" = jasna karnacja * "mysi zapach" => obecność kwasu ortohydroksyfenylooctowego w moczu pacjenta.

Fenyloketonuria Warunkiem pozytywnych efektów - wprowadzenie diety w okresie noworodkowym. Podstawą diety białkozastępcze preparaty nisko- lub bezfenyloalaninowe produkowane na bazie hydrolizatów białkowych lub pozbawione fenyloalaniny syntetyczne mieszaniny aminokwasów uzupełniane w witaminy, składniki mineralne, pierwiastki śladowe.
 u dziewcząt do wieku dorosłego i przez cały")
Fenyloketonuria Ważne kontynuowanie leczenia (diety niskofenyloalaninowej) u dziewcząt do wieku dorosłego i przez cały okres prokreacji. Dziecko urodzone przez matkę chorą na PKU która nie zachowuje niskiego poziomu fenyloalaniny jest narażone na : wady serca, mikrocefalię, upośledzenie umysłowe.

Metabolizm steroidów
 • AR • mutacje w genie reduktazy 7 -dehydrocholesterolu (DHCR 7)")
Zespół Smitha-Lemlego-Opitza (SLOS) • AR • mutacje w genie reduktazy 7 -dehydrocholesterolu (DHCR 7) • Cholesterol - synteza błon komórkowych, hormonów steroidowych istoty białej mózgu • Częstość: 1: 10, 000 to 30, 000; w Polsce, Słowacji, Czechach najwyższa częstość • Objawy: liczne wady rozwojowe OUN (małogłowie), serca, zewnętrznych narządów płciowych, rozszczep podniebienia, zaćma, blepharoptoza, dodatkowe palce, syndaktylia • Zahamowanie wzrostu, upośledzenie rozwoju psychofizycznego, zaburzenia zachowania
 rozszczep podniebienia obojnacze narządy płciowe polidaktylia i bruzda poprzeczna syndaktylia drugiego")
Zespół Smitha-Lemlego-Opitza (SLOS) rozszczep podniebienia obojnacze narządy płciowe polidaktylia i bruzda poprzeczna syndaktylia drugiego i trzeciego palca
")
Upośledzona aktywność 7 - redukatazy 7 -dehydrocholesterolu, która katalizuje przemianę 7 -dehydrocholesterolu (7 -DHC) do cholesterolu i 7 dehydrocholesterolu do desmosterolu. Zahamowanie syntezy cholesterolu i zwiększone stężenie 7 DHC w surowicy.
 Badania biochemiczne krwi – podwyższone stężenie 7 DHC Poziom cholesterolu w")
Zespół Smitha-Lemlego-Opitza (SLOS) Badania biochemiczne krwi – podwyższone stężenie 7 DHC Poziom cholesterolu w surowicy krwi niski, ale u 10% mogą być wartości prawidłowe Analiza mutacji genu DHCR 7, korelacja fenotyp-genotyp Badania w kierunku SLOS powinny być rozważone u płodów/dzieci z polidaktylią, syndaktylią, zaćmą, rozszczepem podniebienia, obojnaczymi narządami płciowymi i prawidłowym kariotypem.

Metabolizm monosacharydów

GALAKTOZEMIA • Defekt metabolizmu galaktozy • częstość 1: 60, 000; AR • Mutacje w genach: galaktozo-1 fosfourydylotransferazy (GALT), GALT UDP-galactozo-4 -epimerazy (GALE) GALE i galaktokinazy 1 (GALK 1) GALK 1 • Homozygota G/G GALT – aktywność enzymu poniżej 5% wartości prawidłowych • Heterozygota G/N – aktywność enzymu około 50% wartości prawidłowych

GALAKTOZEMIA Objawy kliniczne mogą wystąpić w kilka godzin po pierwszym karmieniu: wymioty, brak przyrostu masy ciała, żółtaczka, skaza krwotoczna. powiększenie wątroby i śledziony, zaćma We krwi podwyższone stężenie galaktozy, obniżony poziom glukozy. W moczu: galaktozuria, białkomocz, aminoaciduria (obecność substancji redukujących)

GALAKTOZEMIA Leczenie: całkowite wykluczenie z diety galaktozy i laktozy.

Metabolizm sfingolipidów

Choroba Gauchera • brak glukocerebrozydazy • enzym aktywny w lizosomach, rozkłada glukocerebrozydy do glukozy i ceramidów • glukocerebrozydy obecne są w wielu tkankach, przede wszystkim w makrofagach szpiku, wątroby i śledziony • glukocerebrozydy powstają w procesie rozkładu krwinek czerwonych, fagocytowanych i degradowanych przez makrofagi. • Gromadzenie glukocerebrozydów w lizosomach upośledza funkcje makrofagów. • Makrofagi + nierozłożone glukocerebrozydy - komórki Gauchera

Komórki Gauchera „łagodne” jądra komórkowe i cytoplazma o wyglądzie pogniecionego papieru

Choroba Gauchera • Typ I – dorosłych Hepatosplenomegalia Cytopenia Choroba płuc Choroba kości (patologiczne złamania, bóle stawów i kości) • Typ II – niemowlęca (nie obejmuje kości) między 3 -6 mż słabe przyrosty masy ciała, hepatosplenomegalia, regresja rozwoju, wzmożone napięcie mięśniowe, drgawki, nawracające infekcje ukł. oddechowego, zgon w drugim roku życia

Choroba Gauchera - diagnostyka • Test fluorometryczny – ocena aktywności glukozylocrebrydazy w leukocyach krwi obwodowej wynik nie umożliwia przewidzenia ciężkości ani typu choroby • Analiza mutacji potwierdza rozpoznanie kliniczne i wynik badań biochemicznych • Badanie nosicielstwa w rodzinie probanda

Choroba Gauchera - postępowanie • • • Splenektomia Transfuzja krwi - niedokrwistość Leczenie p-bólowe Leczenie ortopedyczne Suplementacja bifosfonianów i wapnia – metabolizm kości. • Enzymatyczna terapia zastępcza ( (Cerezyme ®), recombinowana glucozyloceramidaza zawierająca alphamannozyl , lepsza fagocytoza przez makrofagi Regularne podania iv zmniejszyło nasilenie objawów hematologicznych, hepatosplenomegalię. Lek dobrze tolerowany Ok. 10 -15% pacientów produkuje p-ciała p-enzymowi Enzymatyczna terapia zastępcza choroba Fabryego, MPSI; Próby kliniczne choroba Pompego, MPS II, VI

WRODZONE WADY METABOLIZMU POSTĘPOWANIE • Ograniczenia dietetyczne metabolizm aminokwasów • Witaminy np. , suplementacja thiaminy w kwasicach melczanowych • Dializa • Enzymatyczna terapia zastępcza • Terapia genowa • Transplantacja narządów, szpiku • Leczenie objawowe • Diagnostyka prenatalna, preimplanatacyjna

Diseases Treated - Inborn Errors of Metabolism Adrenoleukodystrophy Bare-lymphocyte syndrome Dyskeratosis congenita Familial erythrophagocytic lymphohistiocytosis Gaucher disease Gunter disease Hunter syndrome Hurler syndrome (genetic) Inherited neuronal ceroid lipofuscinosis Krabbe disease Langerhans’-cell histiocytosis Lesch-Nyhan disease Leukocyte adhesion deficiency Osteopetrosis - genetic
- Slides: 46