What geneticists do From BaxevanisOuellette p 142 Most

What geneticists do: From Baxevanis/Ouellette, p. 142: Most researchers utilize genome information in one of three ways: • To find out what genomic elements are contained within a genomic region • To determine the order of defined elements within a region • To determine the chromosomal position of a particular element

A Timeline of The Human Genome # human genes mapped to a chromosome location # years it would take to sequence the human genome • 1967 none • 1977 3 genes mapped 4, 000 years to finish at 1977 rate • 1987 12 genes mapped • 1997 30, 000 genes mapped 50 years to finish sequencing not possible yet 1000 years to finish at 1987 rate

Finding Genes that cause Disease? • Does completion of the HGP signal the end of the ‘gene hunt’? • Will the completion of the human genome sequencing lead to the immediate identification of all disease genes?

The STS map: • STS = sequence-tagged site. • STS are short, unique fragments of DNA generated by PCR. • Verification of a human STS: PCR amplification of the human genome generates one small fragment unique landmark.

Usefulness of STSs are used in various mapping activities: • STSs are used to find overlaps between fragments of genomic DNA. • Finding overlaps ordering of fragments (see handout).

Utility of STSs: • STSs would be more valuable if they were assigned to chromosomes and ordered. • STSs are often ordered by Radiation Hybrid Mapping.
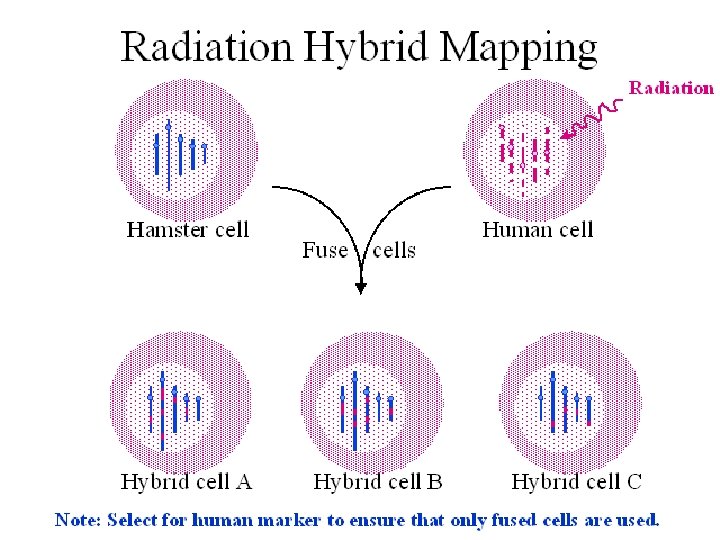

Characteristics of an RH panel • The complete donor genome is represented multiple times in each RH panel • Each hamster cells has numerous fragments of human DNA (~~~ 5 -10 MB).

• Radiation Hybrid Mapping Information Page
 PCR amplify STS in each hybrid (fusion cell) in the")
Ordering/Mapping STSs • 1) PCR amplify STS in each hybrid (fusion cell) in the RH panel. The results is scored as either + (STS present) or – (STS absent). • 2) Data is entered into a central database and pattern matching software generates spatial relationship.
 that physically lie near each other")
Ordering/Mapping STSs • Two markers (STS or other) that physically lie near each other will show similar patterns of retention or loss. • New STSs are mapped by comparing the pattern of positives and negatives with the patterns in the database.

Assigning Chromosomal Location

• Chr 7: Radiation Hybrid Map
 As of Dec. 2001, the ~10 million EST records comprised")
Expressed Sequence Tags (ESTs) As of Dec. 2001, the ~10 million EST records comprised ~72% of the sequences in Gen. Bank. ESTs from mouse and human genomes total over 6 million. Although all of the original ESTs were of human origin, NCBI’s EST database (db. EST) now contains ESTs from over 250 organisms.

What is an EST? Short DNA sequence representing a gene expressed in a particular tissue. A given EST often represents a fraction of the gene. Question- How do you know that the DNA sequence is an expressed sequence?

Generation of an EST • Generation of an EST is initiated by isolation of m. RNA from a tissue of interest. • The m. RNA is converted to a double-stranded c. DNA (complementary DNA) by the viral enzyme reverse transcriptase. • Both ends of the c. DNA (usually) are sequenced. These short sequences are the 5’ EST and the 3’ ESTs!

Human ESTs: • There are ~ 4 million human ESTs in Gen. Bank. . .

What is the value of ESTs? • Rapid identification of genes. Feb. 1992 - Craig Venter and 14 co-workers published the partial DNA sequence of of 2, 375 genes expressed in the human brain. This represented about half of the total human genes known at the time.
 Quickly- focus on the genes")
How to sequence a genome? ? ? • 1) Quickly- focus on the genes and their regulatory regions and human polymorphisms. • 2) Thoroughly and completely- every nucleotide with 99. 99% accuracy.

Big Deal? • Venter and co-workers found novel human genes that show strong sequence similarities to interesting genes from other species. • Fact- Researchers hunting for a novel gene are much more likely to find it in db. EST than in the rest of Gen. Bank.
 applied for patents on")
Patenting of partial gene sequences? ? • NIH (Venter’s employer) applied for patents on approximately 7, 000 partial gene sequences. • Axel Kahn- “I compare this information to the discovery of celestial galaxies. I would patent the moon!”

‘Transcript Map’ • STSs derived from known genes and ESTs ‘Transcript Map’. • (The assignment of expressed sequences to specific chromosome regions is called transcriptional mapping. )

Gene. Map’ 99 • NCBI description: - physical map of >35, 000 human gene-based markers, constructed by the International Radiation Hybrid Mapping Consortium using a consistent set of RH reagents and methodologies. Provides a framework for accelerated sequencing efforts by highlighting key landmarks (gene-rich regions) of the chromosomes, and represents the cooperative efforts of more than one hundred scientists throughout the world.

What is an ‘Integrated Map’?

Map Integration • Map intergration is cross-referencing between various types of maps. • This process in more difficult than it sounds as the units of distance are different and do not exactly translate (1 c. R 3000 = ~ 100 kb).

• Gene. Map'99
 is the official repository for genomic mapping data created by")
The Genome Database (GDB) is the official repository for genomic mapping data created by the Human Genome Project. • GDB stores and curates data generated by researchers engaged in the mapping effort of the HGP. At present, GDB comprises descriptions of the following types of objects: • Regions of the human genome, including genes, clones, amplimers (PCR markers), breakpoints, cytogenetic markers, fragile sites, ESTs, syndromic regions, contigs and repeats. • Maps of the human genome, including cytogenetic maps, linkage maps, radiation hybrid maps, content contig maps, and integrated maps. These maps can be displayed graphically via the Web. • • Variations within the human genome including mutations and polymorphisms, plus allele frequency data.

• The Genome Database

SNPs = single nucleotide polymorphisms • Estimated number- every 500 or 1, 000 nucleotides. Generally thought to be biallelic. • (mutation vs. polyporphism? )

Finding Disease Genes • de. CODE genetics • Why Iceland?

Searching for Disease Genes almost always start with the DNA of affected individuals. Process at Decode Genetics: • 1) Identify people with a particular disease • 2) Find affected people who are related in such a way that they are likely to share genes ( pedigree) • 3) Extract the DNA of these individuals • 4) PCR amplify (robotically) SNPs along each person’s chromosomes • 5) Look for clusters of SNPs among the DNA of patients from a single family. • 6) Such clusters suggest ?

Such clusters suggest a gene involved in the disease is located nearby. • Big deal? • If this data is correct, the gene has now been linked to a particular chromosomal region. Presumably the gene will soon be found. • Where do you go next? ?
")
• Sequenom: From Code to Cure • (see about us)

• Sequenom's scientists are interested in changes in the frequency of SNPs as the population ages. "We take advantage of the fact that most human diseases are late-onset. Age is a major risk factor. . . ”

“If young people are carrying a harmful variation, they're still well, whereas an old person carrying that same variation has a very high chance that he's been made sick or killed by it. You make the prediction that variations that are harmful to health should decline in frequency as a function of age in the healthy population. ”
: • -Drugs target proteins • ~500 known target")
Charles Cantor (C. E. O. Sequenom): • -Drugs target proteins • ~500 known target found in the last century • Next 2 years- ~ all impt. targets identified
![Charles Cantor (C. E. O. Sequenom): • Needed: • a) markers [lots] • b)](http://slidetodoc.com/presentation_image_h/37d2e46158064786dc65138cf7d70f69/image-38.jpg "Charles Cantor (C. E. O. Sequenom): • Needed: • a) markers [lots] • b)")
Charles Cantor (C. E. O. Sequenom): • Needed: • a) markers [lots] • b) populations [big] • c) accurate precise tools [~30% of SNPs are real]
: • Markers- SNPs- (Sequenom has 400, 000 SNP")
Charles Cantor (C. E. O. Sequenom): • Markers- SNPs- (Sequenom has 400, 000 SNP assays that are working). • Population- Sequenom has recruited 15, 000 blood donors • Assay- MALDI-TOF (Matrix-Assisted Laser Desorption Ionization – Time of Flight). Derivative of Mass Spec. Automated, high throughput, accurate.
: • PCR assay involves 3 dd. NTPs, and")
Charles Cantor (C. E. O. Sequenom): • PCR assay involves 3 dd. NTPs, and 1 d. NTPs: • _|||||||||||||||||____ A • _|||||||||||||||||____ G
: • Identified 81 target genes*. * Initially the")
Charles Cantor (C. E. O. Sequenom): • Identified 81 target genes*. * Initially the disease associated with the target gene is unknown. Next Step- Twin Studies
: • Results testing? ? Result of test- AA,")
Charles Cantor (C. E. O. Sequenom): • Results testing? ? Result of test- AA, AG, or GG (crystal clear) Implications of the test- unclear in many cases. Contrast HD testing with HIVR testing
: • treatment? • Pharmacogenomics? ? : • Pharmacogenomics")
Charles Cantor (C. E. O. Sequenom): • treatment? • Pharmacogenomics? ? : • Pharmacogenomics is the study of how an individual's genetic inheritance affects the body's response to drugs.

Jobs at Sequenom? • Sequenom: From Code to Cure

The End
- Slides: 45