VXRIT02 00086 Nov 2015 Eziologia della Fibrosi Cistica

VXR-IT-02 -00086 – Nov 2015
 Il difetto di base a livello cellulare è una")
Eziologia della Fibrosi Cistica (FC) Il difetto di base a livello cellulare è una disfunzione della proteina CFTR* Il gene CFTR codifica una proteina – la proteina-canale CFTR La perdità dell’attività della proteina CFTR deriva di mutazioni del gene CFTR e questo risulta in riduzione della quantità e/o funzione della proteina CFTR alla superficie apicale della cellula Questo risulta in anomalie nel trasporto di ioni nel polmone, pancreas, tratto intestinale, sinus, cute e sistema riproduttivo, provocando I sintomi della FC Rowe SM et al. N Engl J Med 2005; 352: 1992– 2001; Mac. Donald KD et al. Paediatr Drugs 2007; 9: 1– 10 *Cystic fibrosis transmembrane conductance regulator
 è una malattia multisistemica Le manifestazioni multisistemiche sono dovute ad")
La fibrosi cistica (FC) è una malattia multisistemica Le manifestazioni multisistemiche sono dovute ad un difetto di trasporto ionico attraverso la proteina CFTR mutata Sinuses Nasal congestion Loss of smell Chronic infection Nasal polyps Pancreatic insufficiency Fat malabsorption Vitamin deficiency Acute/chronic pancreatitis Gastrointestinal Tract Nutrient malabsorption Gastroesophageal reflux disease Distal intestinal obstructive syndrome (DIOS) Biliary duct obstruction Focal biliary cirrhosis O’Sullivan BP & Freedman SD. Lancet 2009; 373: 1891– 904 Sweat Glands Excessive salt loss Dehydration Chronic metabolic alkalosis Heat prostration Lungs Chronic airway infection, progressing to bronchiectasis, gas trapping, hypoxemia, and hypercarbia Reproductive System Infertility Congenital bilateral absence of vas deferens (CBAVD)

FC - mediana di età nella seconda decade di vita Dati del Registro dei pazienti US 20141 In Italia, la mediana di età era di 29 anni nel periodo 2006 -20112 1. Registro dei pazienti US 2014; 2. Colombo et al, J Cystic Fibrosis 2015; 14: 267 -14

Cascata fisiopatologica della FC E trattamenti attualmente in uso Riassunto delle caratteristiche del prodotto; Welsh MJ et al. Cystic fibrosis: membrane transport disorders. In: Valle D, Beaudet A, Vogelstein B et al, eds. The Online Metabolic & Molecular Bases of Inherited Disease. The Mc. Graw-Hill Companies Inc; 2004: part 21, chap 201. www. ommbid. com. ; Moskowitz SM et al. Genet Med. 2008; 10(12): 851 -868.

KALYDECO – alcune tappe chiave Francis Collins, Lap-Chee Tsui e John R. Riordan scoprono il gene della fibrosi cistica 1989 2004 2006 2012 2014 2015 Ivacaftor è sintetizzato nel centro R&D di Vertex a San Diego Primo studio con Ivacaftor in soggetti con FC Autorizzazione FDA ed EMA di Ivacaftor in soggetti di età pari o superiore a 6 anni con mutazione G 551 D Autorizzazione FDA ed EMA all'uso di Ivacaftor esteso ad altre otto mutazioni «gating» Autorizzazione AIFA all'uso di Ivacaftor in pazienti con una di nove mutazioni «gating»

Prevalencedelle of CFTR Gating Mutations Prevalenza mutazione di Gating Circa 5% della popolazione fibrosi cistica mondiale ha Worldwide, approximately 2, 800 people with CF havedi a gating onsu at una mutazione gating 1 one allele almenoleast 1 allele G 551 D è la mutazione di gating più G 551 Dcon is the most common gating frequente una prevalenza del 4% mutation, with approximately 2, 200 people 1 nel mondo 1 with one allele Le altre mutazioni di account gating for Other gating mutations rappresentano meno dell’ 1%11 approximately 550 patients Other CFTR alleles G 551 D Gating Other gating Based on worldwide estimates. 1. CFF and Johns Hopkins Hospital. Clinical and Functional Translation of CFTR (CFTR 2) Database. http: //www. cftr 2. org/. 7 G 178 R G 551 S G 970 R G 1244 E S 1255 P G 1349 D S 549 N S 549 R S 1251 N

KALYDECO è un potenziatore del CFTR Riassunto delle caratteristiche del prodotto; Welsh MJ et al. Cystic fibrosis: membrane transport disorders. In: Valle D, Beaudet A, Vogelstein B et al, eds. The Online Metabolic & Molecular Bases of Inherited Disease. The Mc. Graw-Hill Companies Inc; 2004: part 21, chap 201. www. ommbid. com.
 N pazienti Metodologia")
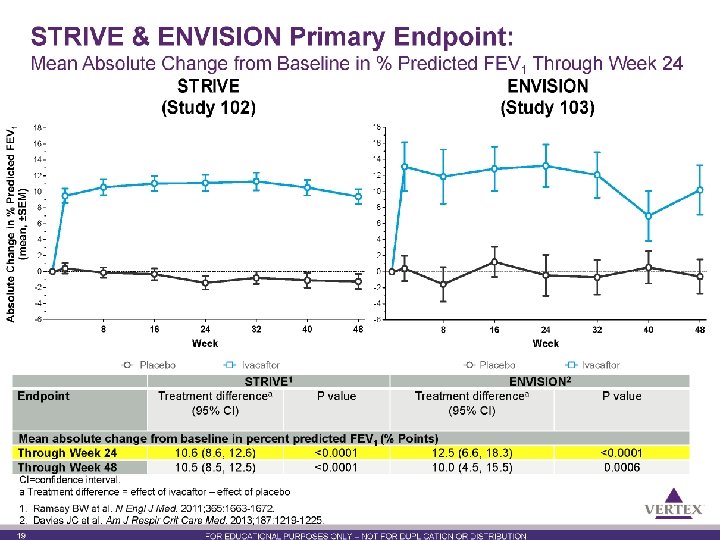
Riassunto degli studi clinici di Fase III Studio Mutazioni Età (anni) N pazienti Metodologia Durata STRIVE G 551 D ≥ 12 167 Doppio cieco vs placebo 48 settimane ENVISION G 551 D 6 -11 52 Doppio cieco vs placebo 48 settimane PERSIST G 551 D ≥ 6 192 Roll over STRIVE/ENVISION in aperto 96 settimane KONNECTION G 1244 E, G 1349 D, G 178 R, G 551 S, S 1251 N, S 1255 P, S 549 N, S 549 R o G 970 R ≥ 6 39 Cross over doppio cieco vs placebo Seguito da periodo in aperto 8 settimane + 16 settimane 9 BW, delle et al. N Engl del J Med. Nov Davies al. Am J Respir Crit Care Med. 2013 Jun 1; 187(11): 1219 -25 1. Ramsey Ivacaftor, Riassunto Caratteristiche Prodotto. 2011 2. Ramsey BW, 3; 365(18): 1663 -72; et al. ; VX 08 -770 -102 Study Group. N Engl. JC, J Med. et 2011; 365(18): 1663 -72. TM 3. Mc. Kone EMA-Agenzia Europea Relazione pubblica europea di valutazione (EPAR) per Kalydeco; De 2012; Comitato medicinali per uso umano EMA/473279/2012. EF et dei al, Farmaci. Lancet Respir Med. 2014 Nov; 2(11): 902 -10. Boeck K, pereti prodotti al. J Cyst Fibros. 2014(CHMP), Dec; 13(6): 674 -80

10

11

Riassunto degli studi clinici di Fase III In pazienti con mutazioni di gating di età superiore ai 6 anni, KALYDECO ha dimostrato, rispetto al placebo (standard of care) • Miglioramento rapido e costante della funzione polmonare • Riduzione delle esacerbazioni polmonari • Miglioramento dello stato nutrizionale evidenziato dall’aumento del peso corporeo / BMI • Riduzione della concentrazione del cloro sudorale • Incidenza di eventi avversi simile al placebo Riassunto delle caratteristiche del prodotto 1. Ivacaftor, Riassunto delle Caratteristiche del Prodotto. 2. Ramsey BW, et al. ; VX 08 -770 -102 Study Group. N Engl J Med. 2011; 365(18): 1663 -72. 3. EMA-Agenzia Europea dei Farmaci. Relazione pubblica europea di valutazione (EPAR) per Kalydeco TM 2012; Comitato per i prodotti medicinali per uso umano (CHMP), EMA/473279/2012. 12

Kalydeco è indicato per il trattamento della fibrosi cistica in pazienti di età pari o superiore a 6 anni che hanno una delle seguenti mutazioni di gating (di classe III) nel gene CFTR: G 551 D, G 1244 E, G 1349 D, G 178 R, G 551 S, S 1251 N, S 1255 P, S 549 N o S 549 R.


VXR-IT-02 -00086 – Nov 2015

Le mutazioni di CFTR possono ridurre la quantità o la funzione della proteina CFTR sulla superficie cellulare Mutazione di CFTR Ridotta quantità di CFTR sulla superficie cellulare Normale Ridotta funzione di CFTR sulla superficie cellulare Difetto di processamento o sintesi Difetto di gating Difetto di conduttanza (Classi 1, 2 e 5) (Classe 3) (Classe 4) Mac. Donald KD et al. Pediatr Drugs. 2007; 9: 1 -10. Zielenski J. Respiration. 2000; 67: 117 -133. Welsh MJ et al. Cystic fibrosis. In: Valle D et al, eds. OMMBID. The Mc. Graw-Hill Companies Inc; 2004: part 21, chap 201.

L'approccio terapeutico di Vertex consiste nell'aumentare la quantità e la funzionalità di CFTR sulla superficie cellulare Facilitare il trasporto di cloro aumentando la quantità di CFTR che raggiunge la superficie cellulare Facilitare il trasporto di cloro potenziando la probabilità di apertura del canale (o gating) della proteina CFTR sulla superficie cellulare es. Ivacaftor (VX-770) Attività totale di CFTR = Densità superficiale * Probabilità di apertura * Conduttanza Zielenski J. Respiration 2000; 67: 117– 33; Mac. Donald KD et al. Paediatr Drugs 2007; 9: 1– 10; Boyle MP & De Boeck K. Lancet Respir Med 2013; 1: 158– 63; Welsh MJ & Smith AE. Cell 1993; 73: 1251– 4; Castellani C. J Cyst Fibros 2008; 7: 179– 96

Ivacaftor aumenta la probabilità di apertura del canale G 551 D-CFTR espresso in colture cellulari Ivacaftor Trasporto totale di cloro = Quantità sulla superficie cellulare Apertura X del canale CFTR normale Aperto Chiuso X Conduttanza del canale G 551 D-CFTR Aperto Chiuso 1 p. A G 551 D-CFTR più Ivacaftor 3 sec Aperto Chiuso Van Goor et al. , Proc Natl Acad Sci U S A. 2009 Nov 3; 106(44): 18825 -30.
")
STRIVE & ENVISION: Study Designs Primary Analysis STRIVE 1 Screening + Randomization (1: 1) − 14 Day − 35 Ivacaftora 150 mg q 12 h Placebo 0 Week 24 Open-label rollover study Ivacaftora 150 mg q 12 h Or 2 -year follow-up 48 Primary Analysis ENVISION 2 Part A Single-dose PK trial Part B Randomization (1: 1) Part B enrollment optional Day − 35 a In − 14 Ivacaftora 150 mg q 12 h Placebo 0 Ivacaftora 150 mg q 12 h Or 2 -year follow-up 48 addition to prescribed CF therapies. The use of inhaled hypertonic saline was not permitted. 1. Ramsey BW et al. N Engl J Med. 2011; 365: 1663 -1672. 2. Davies JC et al. Am J Respir Crit Care Med. 2013; 187: 1219 -1225. 19 Week 24 Open-label rollover study

21

22

23

KONNECTION Study Design D = day; W = week; q 12 h = every 12 hours. • Key eligibility criteria - Diagnosis of CF, patients ≥ 6 years of age - A non-G 551 D-CFTR gating mutation (G 178 R, S 549 N, S 549 R, G 551 S, G 970 R, G 1244 E, S 1251 N, S 1255 P or G 1349 D) on at least one allele but no G 551 D mutations on either allele - FEV 1 ≥ 40% of predicted at screening De Boeck K, et al. J Cyst Fibros. 2014 Dec; 13(6): 674 -80

Mean Absolute Change From Baseline in Percent Predicted FEV 1: 8 Weeks Treatment difference through Week 8 by Mixed Effects Model for Repeated Measures: 10. 7 % points (P<0. 0001) n=37 n=38 vv n=37 De Boeck K, et al. J Cyst Fibros. 2014 Dec; 13(6): 674 -80 v FEV 1 = forced expiratory volume in 1 second; SE = standard error. Observed (raw) mean changes from baseline are plotted at each time point.

Mean Absolute Change From Baseline in BMI: 8 Weeks Treatment difference at Week 8 by Linear Mixed Model: 0. 7 kg/m 2 (P<0. 0001) n=37 n=38 vv n=37 BMI = body mass index; SE = standard error. Observed (raw) mean changes from baseline in BMI are plotted at each time point. De Boeck K, et al. J Cyst Fibros. 2014 Dec; 13(6): 674 -80 n=37 v n=37

Mean Absolute Change From Baseline in Sweat Chloride: 8 Weeks Treatment difference through Week 8 by Mixed Effects Model for Repeated Measures: -49. 2 mmol/L (P<0. 0001) n=36 n=35 n=36 vv n=33 n=34 n=36 v SE = standard error. Observed (raw) mean changes from baseline in sweat chloride are plotted at each time point. De Boeck K, et al. J Cyst Fibros. 2014 Dec; 13(6): 674 -80

Ivacaftor rallenta il tasso di perdita progressiva di funzionalità polmonare FEV 1 percentuale stimata del predetto Miglioramento iniziale Gruppo ivacaftor G 551 D Gruppo di controllo F 508 de Ridotto tasso di declino, P=0, 02 8, 13 punti 9, 05 punti 9, 96 punti 10, 88 punti Inizio dell'analisi *P<0, 001. Stimato con equazioni di Knudson. Anno 1 Anno 2 Anno 3 Basale FEV 1 percentuale stimata del predetto nel Gruppo Ivacaftor G 551 D e nel Gruppo di Controllo F 508 del a 3 anni sulla base di un modello di pazienti trattati con Ivacaftor negli studi clinici ed appaiati per propensity score a controlli del Registro dei Pazienti della Cystic Fibrosis Foundation • Il tasso annuo medio stimato errore standard (ES) della variazione del FEV 1 percentuale del predetto (calcolato con le equazioni di Knudson 2) è stato di − 0, 81 0, 36 punti percentuali nel gruppo ivacaftor G 551 D rispetto a − 1, 73 0, 17 punti percentuali nel gruppo di controllo F 508 del (P=0, 02) e fornisce quindi l'evidenza del fatto che ivacaftor modifica il corso del declino della funzionalità polmonare in pazienti con FC 1. Sawicki GS, et al. J Cyst Fibros 2014; 13(suppl 2): S 6. 2. Knudson RJ, et al. Am Rev Respir Dis 1976; 113: 587 -600. Presentazione alla 28° Conferenza Annuale Nordamericana sulla Fibrosi Cistica (NACFC), Atlanta, GA, USA, 9 -11 ottobre 2014 Sawicki GS, et al, American Journal of Respiratory and Critical Care Medicine, 2015, Vol. 192: 836 -842

Gran parte delle mutazioni responsabili della FC sono estremamente rare France Canada 1: 2, 500 F 508 del 621+1 G T G 542 X United Kingdom 1: 2, 400 F 508 del G 551 D R 117 H USA 1: 3, 500 F 508 del G 542 X G 551 D Mexico 1: 8, 500 F 508 del G 542 X S 549 N 1: 4, 700 F 508 del G 542 X N 1303 K Russian Federation 1: 4, 900 F 508 del CFTRdele 2, 3(21 kb) N 1303 K Germany 1: 3, 300 F 508 del N 1303 K G 542 X • Solo 5 mutazioni hanno una frequenza >1% • ~20 mutazioni Ireland hanno una frequenza >0. 1% 1: 1, 400 • F 508 del èF 508 del la mutazione più comune presente su G 551 D in ~90% dei soggetti con CF almeno 1 allele R 117 H Brazil 1: 6, 902 F 508 del G 542 X R 1162 X The 3 most common CFTR mutations in each country are shown CFTR 2 database. http: //cftr 2. org/index. php; local registries Spain 1: 3, 700 F 508 del G 542 X 1811+1. 6 kb. A G South Africa 1: 7, 056 F 508 del 3120+1 G A G 542 X Japan 1: 100, 000– 1: 350, 000 1540 del 10 125 G C F 508 del India 1: 40, 000– 1: 100, 000 F 508 del 3849+10 kb. C T 3601 -20 T C Australia 1: 2, 900 F 508 del G 551 D G 542 X New Zealand 1: 3, 200 F 508 del G 551 D G 542 X

Popolazione di pazienti con fibrosi cistica Sviluppo di nuovi modulatori del CFTR Sviluppi futuri per la cura della fibrosi cistica G 551 D & Gating Età: 6+ IVACAFTOR

31
- Slides: 31