UNIT I Protein Structure and Function Structure of






 is written to")


















 • • • Named so because they often connect successive")

 • Globular proteins constructed by combining 2º structural elements (α-helices,")











 in side chains of")















- Slides: 57
UNIT I: Protein Structure and Function Structure of Proteins
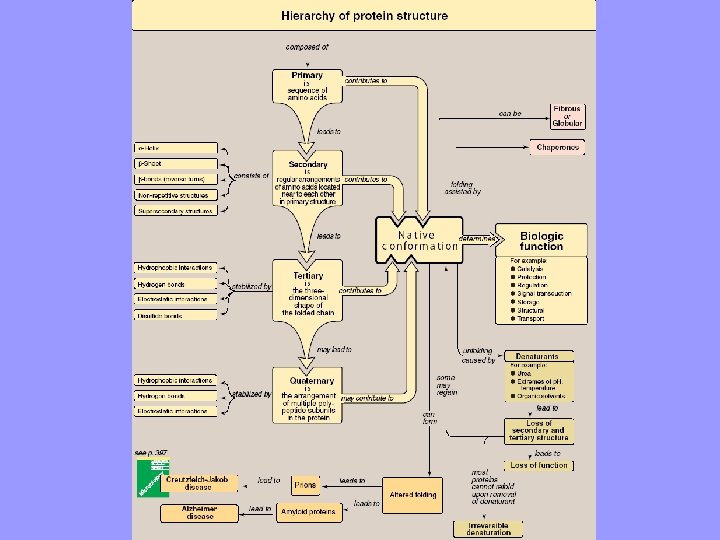
I. Overview • The 20 common aa’s joined by peptide bonds • Linear sequence of linked aa’s info. to generate a protein molecule with a unique 3 D • Complexity of protein molecules analyzed in terms of 4 organizational levels: 1º, 2º, 3º and 4º • Analyses revealed that certain structural elements are repeated in various proteins suggesting general rules followed in protein folding • Repeated structural elements range from simple αhelices and β-sheets forming small motifs to complex folding of polypeptide domains of multifunctional proteins
Figure 2. 1. Four hierarchies of protein structure.
II. Primary structure of proteins • The sequence of aa’s in a protein is called 1º structure of protein • Many genetic diseases result in proteins with abnormal aa sequences, causing improper folding and loss or impairment in function • If normal and mutant aa sequences known, info. may be used to diagnose or study disease
A. Peptide bond • In proteins aa’s are joined covalently by peptide bonds, i. e. , amide linkages b/w αcarboxyl of one aa and α-amino group of another. e. g. , valylalanine. Figure 2. 2 -A. Formation of a peptide bond, showing the structure of the dipeptide valylalanine.
• Peptide bonds are not broken by conditions that denature proteins, e. g. , heating or high conc. of urea. • Prolonged exposure to a strong acid or base + elevated temp. is required to hydrolyze nonenzymatically.
1. Naming peptide: the free amino end of peptide chain (N-terminal) is written to left and free carboxyl end (C-terminal) to the right. • • • Linkage of many aa’s through peptide bonds unbranched chain (= polypeptide) Each aa in a polypeptide is called a “residue” or “moiety” When named, aa residues have their suffixes (-ine, -an, -ic, or –ate) changed to –yl, except C-terminal aa, e. g. , a tripeptide of N-terminal Val, Gly, C-terminal Leu is: valylglycylleucine.
2. Characteristics of the peptide bond: It has a partial double-bond character, i. e. , it is shorter than a single bond, and is rigid and planar. • This prevents free rotation around the bond b/w the carbonyl carbon and the nitrogen of the peptide bond • The bonds b/w the α-carbons and α-amino or αcarboxyl groups can be freely rotated (but limited by size and character of the R-groups) allowing polypeptide chain to assume a variety of possible configurations • The peptide bond is generally a trans bond, mainly because of steric interference of R-groups when in cis position
Figure 2. 2 -B. Characteristics of the peptide bond.
3. Polarity of the peptide bond: Like all amide linkages, the –C=O and –NH groups of peptide bond are uncharged, and neither accept nor release protons over p. H 2 -12. • • Thus charged groups in a polypeptide consist of N-terminal α-amino, C-terminal αcarboxyl, and any ionized groups in side chains. The –C=O and –NH groups of peptide bond are polar, and involved in H-bonds, e. g. , in α -helices and β-sheet structures
B. Determination of amino acid composition of a polypeptide • First step in determining 1º structure of a polyp. is to identify and quantitate its aa’s. • A purified sample of a polyp. to be analyzed is hydrolyzed by strong acid at 110 ºC for 24 h. – Treatment cleaves peptide bonds and releases free aa’s separated by cation-exchange chromatography. – The aa’s bind to column with different affinities, depending on their charges, hydrophobicity and other characteristics – Each aa is released from column by eluting with solutions of increasing ionic strength and p. H – Separated aa’s quantitated by heating them with ninhydrin (forms purple cpd with most aa’s, ammonia, and amide), amount of each aa is determined spectrophotometrically by measuring amount of light absorbed by ninhydrin derivatives. – Analysis described is performed by an automated aa analyzer
Figure 2. 3. Determination of the amino acid composition of a polypeptide using an amino acid analyzer.
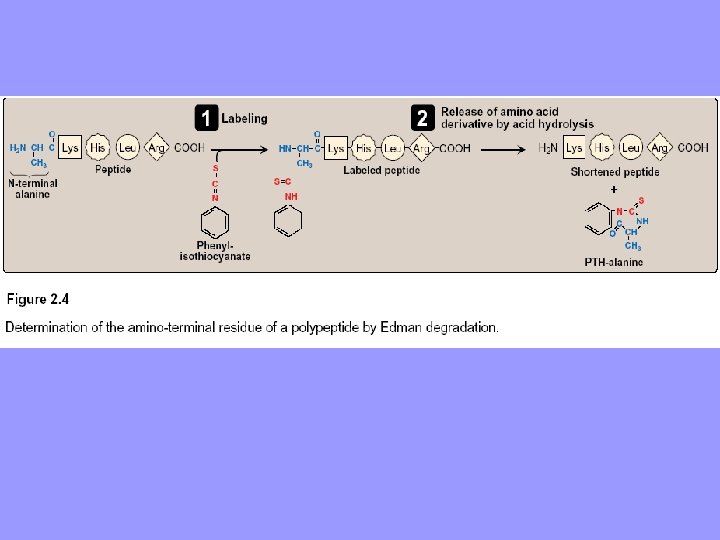
C. Sequencing of the peptide from its N-terminal end • Sequencing: identifying the specific aa’s at each position in the polyp. beginning at N-terminal end. • Phenylisothiocyanate, a. k. a Edman’s reagent, used to label N-terminal residue under mildly alkaline conditions. • Resulting phenylthiohydantoin (PTH) derivative introduces an instability in N-terminal peptide bond selectively hydrolyzed without cleaving other peptide bonds. • Identity of aa derivative determined • Edman’s reagent applied repeatedly to each shortened polyp. obtained in previous cycle • Process automated, and repetition is employed by a machine (sequenator) to determine sequence of > 100 aa’s, starting at N-terminal of a polyp.
D. Cleavage of polypeptide into smaller fragments • • • Many polyp. composed of > 100 aa’s cannot be sequenced directly by a sequenator Large polyps. cleaved at specific sites fragments sequenced Using more than one cleaving agent (enzymes and/or chemicals) on separate samples of purified polyp. overlapping fragments permit proper ordering of sequenced fragments complete aa sequence of a large polyp.
Figure 2. 5. Overlapping of peptides produced by the action of trypsin and cyanogen bromide.
E. Determination of a protein’s primary structure by DNA sequencing • • • Seq. of nucleotides in a coding region of DNA specifies aa seq. of a polyp. If nucleotide seq. determined from genetic code nucleotide seq. translated aa seq. of polyp. This method cannot predict positions of disulfide bonds in the folded chain Cannot identify any aa modified after incorporation into polyp. (post-translational modification) Direct protein sequencing extremely important tool for determining true character of 1º sequence of many polyps.
III. Secondary structure of proteins • Polyps generally form regular arrangements of aa’s that are located near each other in the linear sequence • These arrangements are called 2º structures of polyp. • α-helix, β-sheet, and β-bend are examples of 2º structures encountered in proteins • The collagen helix is another example of 2º structure
A. α-Helix • Several different polyp. helices, α-helix is most common • α-helix is a spiral structure, tightly packed, coiled backbone core, with R-groups extending outward to avoid interfering sterically with each other. • Various proteins contain α-helices. e. g. , keratins are fibrous proteins whose structure is nearly entirely αhelical. – Keratins are a major component of tissues e. g. , hair and skin, and their rigidity is determined by the number of disulfide bonds b/w constituent polyp chains • In contrast, myoglobin, with ~80% α-helical, is a globular, flexible molecule.
1. Hydrogen bonds: An α-helix is stabilized by hydrogen bonding b/w peptide-bond carbonyl oxygens and amide hydrogens – – – The H-bonds extend up the spiral from the carbonyl oxygen of one peptide bond to the –NH- group of a peptide linkage 4 residues ahead in the polyp This ensures that all but first and last peptide bond components are linked to each other through Hbonds H-bonds are individually weak, but collectively serve to stabilize the helix.
Figure 2. 6. α-Helix showing peptide backbone.
2. Amino acids per turn: – Each turn contains 3. 6 aa’s. Thus, aa’s spaced 3 or 4 apart are spatially close together when folded in α-helix.
3. Amino acids that disrupt an α-helix: • • • Proline disrupts α-helix because its imino group is not compatible with the right handed spiral of α-helix. Instead it insert a kink in the chain, which interferes with helical structure Large number of charged aa’s (e. g. , Glu, Asp, His, Lys, Arg) also disrupt α-helix by forming ionic bonds, or by electrostatically repelling each other Amino acids with bulky side chains, e. g. , Trp, or aa’s e. g. , Val, Ile, that branch at the β-carbon can interfere with formation of α-helix if present in large numbers
B. β-Sheet – – – All peptide bond components involved in H-bonding. Surfaces of β-Sheets appear “pleated”, hence “β-pleated sheets” When illustrated, β-strands are expressed as broad arrows Figure 2. 7. A. Structure of a βsheet. B. An anti-parallel β-sheet with the β-strands represented as broad arrows.
1. Comparison of a β-sheet and an α-helix - β-sheets are composed of two or more peptide chains (βstrands), or segments of polyp chains, which are almost fully extended - The H-bonds in β-sheets are perpendicular to the polyp backbone 2. Parallel and antiparallel sheets - A β-sheet can be formed from ≥ 2 separate polyp chains or segments of polyp chains arranged either antiparallel or parallel - When H-bonds are formed b/w the polyp backbones of separate polyp chains = interchain bonds - A β-sheet can be formed by a single polyp chain folding back on itself, here H-bonds = intrachain bonds - In globular proteins, β-sheets always have right-handed curl, or twist, when viewed along the polyp backbone
Figure 2. 7. B. An anti-parallel β-sheet with the β-strands represented as broad arrows. C. A parallel β-sheet formed from a single polypeptide chain folding back on itself.
C. β-Bends (reverse turns) • • • Named so because they often connect successive strands of antiparallel β-sheets β-Bends reverse the direction of a polyp chain, helping it form a compact, globular shape Usually found on surface of protein molecules & often include charged residues Generally composed of 4 aa’s, one of which may be proline. Gly is also frequently found in β-Bends. Stabilized by H-bonds and ionic bonds
D. Non-repetitive secondary structures • About one half of an average globular protein is organized into repetitive structure • The remainder is described as having a loop or coil formation. • These non-repetitive 2º are not “random”, but rather less regular than those described above
E. Super-secondary structures (motifs) • Globular proteins constructed by combining 2º structural elements (α-helices, β-sheets, nonrepetitive) core region i. e. , interior of molecule. • They are connected by loop regions (e. g. , βbends) at surface of protein. • Super-secondary structures are usually produced by packing side chains from adjacent 2º structural elements close to each other • Thus, e. g. , α-helices and β-sheets that are adjacent in the amino acid sequence are also usually adjacent in, folded protein.
Figure 2. 8. Some common structural motifs combining α-helices and β-sheets. The names describe their schematic appearance.
IV. Tertiary structure of globular proteins • 3º refers both to folding of domains (basic units of structure and function), and final arrangement of domains in a polyp. • Structure of globular proteins in aqueous soln is compact, with a high density (close packing) of atoms in core of molecule. • Hydrophobic R-groups buried in interior, hydrophilic groups generally on surface. All hydrophilic groups (including components of peptide bonds) located in interior are involved in H-bonds or electrostatic interactions. • The α-helix & β-sheet structures provide maximal H-bonding for peptide bond components within interior of polyps eliminating possibility that water molecules may become bound to these hydrophilic groups and, thus, disrupt integrity of protein
A. Domains • Fundamental functional and 3 D structural units of a polyp. • Polyps > 200 aa’s in length generally 2 or more domains • Core of domain is built from combinations of super-secondary structural elements (motifs) • Folding of peptide chain within a domain usually occurs independently of folding in other domains • So, each domain has characteristics of a small, compact globular protein that is structurally independent of other domains in polyp chain
B. Interactions stabilizing tertiary structure • Four types of interactions cooperate in stabilizing 3º structures of globular proteins Disulfide bonds: covalent linkage formed from –SH of each 2 cysteine residues to produce cystine residue. 1. – – The 2 Cys residues may be far apart in the 1º sequence or even on 2 different polyps. Folding of polyp chain(s) brings Cys residues into proximity Contribute to stability of 3 D shape of protein e. g. , many disulfide bonds are found in proteins as Ig’s that are secreted by cells These strong covalent bonds prevent proteins from becoming denatured in extracellular environ.
Figure 2. 9. Formation of a disulfide bond by the oxidation of two cysteine residues, producing one cystine residue.
2. Hydrophobic interactions: aa’s with nonpolar R-groups tend to be located in interior of polyp, where they associate with other hydrophobic aa’s – In contrast, polar or charged aa’s tend to be located on surface in contact with polar solvent • For protein located in non-polar (lipid) environ. e. g. , membrane, aa’s exhibit reverse arrangement.
Figure 2. 10 Hydrophobic interactions between amino acids with nonpolar side chains.
Figure 1. 4 Location of nonpolar amino acids in soluble and membrane proteins.
3. Hydrogen bonds: aa R-groups containing oxygen- or nitrogen-bound hydrogen, e. g. alcohol groups of Ser and Thr, can form H-bonds with electron rich atoms e. g. , oxygen of a carboxyl group or carbonyl group of a peptide bond – Formation of H-bonds b/w polar groups on protein surface and aqueous solvent enhances solubility of protein
Figure 2. 11 Interactions of side chains of amino acids through hydrogen bonds and ionic bonds.
Figure 1. 6 Hydrogen bond between the phenolic hydroxyl group of tyrosine and another molecule containing a carbonyl group.
4. Ionic interactions: negatively charged groups, e. g. , (-COO-) in side chains of Asp or Glu can interact with positively charged groups, e. g. , (-NH 3+) in side chain of Lys
C. Protein folding • Interactions b/w side chains of aa’s determine folding of long polyp into 3 D shape of functional protein • Protein folding, occurs within cell in seconds to minutes, employs a shortcut through the maze of all folding possibilities • As a peptide folds, its aa R groups attracted and repulsed according to their chemical properties, e. g. , charged side chains • Plus, interactions involving H-bonds, hydrophobic interactions, and disulfide bonds • This process of trial and error tests many possible configurations, where interactions outweigh repulsions correctly folded protein with a low energy state
Figure 2. 12. Steps in protein folding.
D. Role of chaperones in protein folding • Generally accepted: info needed for correct protein folding is contained in 1º structure of polyp. • Difficult to explain, why most proteins when denatured do not resume native conformation • One answer: a protein begins to fold in stages during its synthesis, rather than waiting completion of synthesis of entire polyp limits competing folding configurations made available by longer stretches of nascent polyp.
• Additionally, a specialized group of proteins “chaperones” required for proper folding of many proteins. – Chaperones, a. k. a. “heat shock” proteins interact with polyp at various stages during folding – Some chaperones are important in keeping polyp unfolded until its synthesis finished, or act as catalysts by increasing rates of final stages in folding process – Others protect proteins as they fold so that their vulnerable, exposed regions do not become tangled in unproductive encounters
V. Quaternary structure of proteins • Many proteins consist of a single polyp. monomeric proteins • Others may consist of ≥ 2 polyp chains that may be structurally identical or totally unrelated • Arrangement of these polyp subunits = quaternary structure ( 2 subunits = dimeric, 3 subunits = trimeric, several subunits = multimeric) • Subunits held together by non-covalent interactions (H-bonds, ionic, hydrophobic) • Subunits may function independently of each other, or cooperate as in Hb binding of oxygen to one subunit of the tetramer increases affinity of the other subunits for oxygen
VI. Denaturation of proteins • Results in unfolding and disorganization of proteins’ 2º and 3º structures, not accompanied by hydrolysis of peptide bonds • Denaturing agents include heat, organic solvents, mechanical mixing, strong acids or bases, detergents and ions of heavy metals e. g. , lead and mercury • Denaturation may under ideal conditions be reversible i. e. , protein refolds into original native structure when denaturing agent removed • Most proteins, once denatured, remain permanently disordered • Denatured proteins are often insoluble and, so precipitate in solution
VII. Protein misfolding • Protein folding is a complex, trial and error process that sometimes improperly folded protein • Misfolded proteins usually tagged and degraded within the cell but this quality control system is not perfect and intracellular or extracellular aggregates of misfolded proteins can accumulate, particularly as individuals age • Deposits of misfolded proteins are associated with a number of diseases including amyloidoses
A. Amyloidoses • Misfolding of proteins may occur spontaneously, or by mutation in a gene which produces altered protein • Plus, some apparently normal proteins abnormal proteolytic cleavage unique conformational state long, fibrillar protein assemblies consisting of β-pleated sheets • Accumulation of these spontaneously aggregating proteins, called “amyloides”, has been implicated in many degenerating diseasesparticularly in the neurodegenerative disorder, Alzheimer disease.
Alzheimer • Dominant component of amyloid plaque that accumulates in Alzheimer is Aβ, a peptide 40 -43 aa’s • X-ray crystallography and IR spectroscopy demonstrate a characteristic β-pleated sheet conformation in nonbranching fibrils • This peptide, when aggregated in β-pleated sheet configuration, is neurotoxic, is central pathogenic event cognitive impairment characteristic of disease • Aβ amyloid deposited in brain in Alzheimer is derived by proteolytic cleavages from the larger amyloid precursor protein – a single transmembrane protein expressed on cell surface in brain and other tissues • Aβ peptides aggregate amyloid found in brain parenchyma and around blood vessels.
Figure 2. 13. Formation of amyloid plaques found in Alzheimer disease.
• Most Alzheimer cases are not genetically based, although 5 -10% of cases are familial • A second biologic factor involved in development of Alzheimer is accumulation of neurofibrillay tangles in brain – A key component of these tangled fibers is an abnormal form of tau protein, which in healthy form helps in assembly of microtubular structure – Defective tau, however, appears to block actions of its normal counterpart
B. Prion disease • Prion protein “Pr. P”, implicated as causative agent of transmissible spongiform encephalopathies (TSE), including: – Creutzfeldt-Jakob disease in humans – Scrapie in sheep – Bovine spongiform encephalopathy “BSE” in cattle (a. k. a “mad cow disease”) • Infectivity of agent causing scrapie was associated with a single protein species that was not associated with detectable nucleic acid • Infectious protein is designated “prion protein”, is highly resistant to proteolytic degradation • When infectious tends to form insoluble aggregates of fibrils, similar to amyloid found in other brain diseases
• A non-infectious form of Pr. P, having same aa and gene sequences as infectious agent, is present in normal brains on surface of neurons and glial cells • i. e. , Pr. P is a host protein. No 1º structure differences or alternate posttranslational modifications b/w normal and infectious forms • The key to becoming infectious lies in changes in 3 D conformation of Pr. P, e. g. , a number of α-helices replaced by β-sheets in infectious form • Presumably, conformational difference relative resistant to proteolytic degradation & permits distinguishing infectious from normal • Infectious agent is an altered version of normal protein, which acts as a “template” for converting normal protein to pathogenic conformation • TSEs are invariably fatal, no treatment is available
Figure 2. 14. One proposed mechanism for multiplication of infectious prion agents.
Summary • Native conformation : functional fully-folded protein structure • 3 D structure is determined by 1º structure (i. e. , aa sequence) • Interactions b/w R-groups guide folding 2º, 3º, and sometimes 4º • A specialized group “Chaperones” required for proper folding of many proteins • Protein denaturation unfolding & disorganization of protein’s structure without hydrolyzing peptide bonds • Denaturation may be reversible or, more commonly irreversible • Disease can occur as a normal protein a conformation that is cytotoxic e. g. , Alzheimer and TSEs • In Alzheimer, normal protein after abnormal processing conformation neurotoxic amyloid protein consisting of βpleated sheets. Additionally, abnormal tau protein • In TSEs infective agent is an altered version of a normal prion protein that acts as a template for converting normal protein to pathogenic conformation