TRASTORNOS DEL METABOLISMO DE LAS BIOPTERINAS 24 enero

TRASTORNOS DEL METABOLISMO DE LAS BIOPTERINAS 24 enero 2014 Dra. Daniela Muñoz Ch. Dra. Diane Vergara G. Neuróloga Infantil Pediátrica Becada Neurología

Introducción Neurotransmisor Clasificación bioquímica Se sintetiza en la neurona Se presenta en la terminal presináptica y se libera en cantidades suficientes para ejercer una acción determinada en la neurona postsináptica Aminas biógenas: catecolaminas e indolaminas Acetilcolina Administrado en cantidades suficientes reproduce la acción de la molécula endógena Existe un mecanismo específico para eliminarse del espacio sináptico Aminoácidos: GABA, glut, NAA, glicina, serina Purinas: AMP, ADP, ATP Neuropéptidos Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 García-Cazorla A et al. Errores congénitos de los neurotransmisores en Neuropediatría. Rev neurol 2005; 41: 99 -108

Enfermedades de neurotransmisores en pediatría Grupo de trastornos neurometabólicos hereditarios atribuibles a una alteración primaria del metabolismo de neurotransmisores (NT) o su transporte Grupo en crecimiento, requiere métodos diagnósticos especiales Primera descripción: convulsiones que responden a piridoxina (1954) En los últimos años expansión del conocimiento respecto las bases moleculares de estas enfermedades, su expresión fenotípica y opciones de tratamiento Síntomas determinados por el tipo y severidad del trastorno � Lo central: manifestaciones neurológicas Importante alto índice de sospecha: tratamiento Pearl PL, et al. Diagnosis and Treatment of Neurotransmitter Disorders. Curr Treat Options Neurol. 2006 Nov; 8(6): 441 -50 Pons R. The phenotypic spectrum of paediatric neurotransmitter diseases and infantile parkinsonism. J Inherit Metab Dis (2009) 32: 321– 332 Campeau PM, et al. Neurotransmitter diseases and related conditions. Mol Genet Metab. 2007 Nov; 92(3): 189 -97 Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33

Enfermedades de neurotransmisores en pediatría: grupos clásicos Vía de aminoácidos Monoaminas o aminas biógenas • Epilepsia severa precoz (glicina) • Epilepsia, trastorno del lenguaje expresivo, trastornos psiquiátricos (GABA) • Amplio espectro de manifestaciones Pons R. The phenotypic spectrum of paediatric neurotransmitter diseases and infantile parkinsonism. J Inherit Metab Dis (2009) 32: 321– 332

Clasificación Pearl PL, et al. Diagnosis and Treatment of Neurotransmitter Disorders. Curr Treat Options Neurol. 2006 Nov; 8(6): 441 -50

TRASTORNOS DE LAS MONOAMINAS

Trastornos de las aminas biógenas o monoaminas Vía incluye síntesis y catabolismo de � Catecolaminas: dopamina, Noradrenalina (NA) y adrenalina � Indoleamina: serotonina Desde tirosina Desde triptófano Son NT importantes en SNC y SNP, también funcionan como hormonas Pons R. The phenotypic spectrum of paediatric neurotransmitter diseases and infantile parkinsonism. J Inherit Metab Dis (2009) 32: 321– 332 Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Trastornos de las monoaminas: vía dopaminérgica Manifestaciones extrapiramidales Vía nigroestriatal Neuronas dopaminérgicas (mesencéfalo: SNc, área tegmental ventral) También en hipotálamo (función vegetativa y hormonal) Control del movimiento voluntario Vía mesolímbica Cognición y comportamiento Vía mesocortical Pons R. The phenotypic spectrum of paediatric neurotransmitter diseases and infantile parkinsonism. J Inherit Metab Dis (2009) 32: 321– 332
 Proyección difusa")
Trastornos de las monoaminas: vía serotoninérgica Neuronas serotoninérgic as (núcleos del rafe) Proyección difusa al cerebro y médula espinal Control apetito, sueño, memoria y aprendizaje, T° corporal, ánimo, comportamiento sexual, función cardiovascular, contracción muscular, homeostasis También en pared de intestino y vasos sanguíneos Pons R. The phenotypic spectrum of paediatric neurotransmitter diseases and infantile parkinsonism. J Inherit Metab Dis (2009) 32: 321– 332
 Proyección")
Trastornos de las monoaminas: vía noradrenérgica y adrenérgica Neuronas noradrenérgica s (locus ceruleus) Proyección difusa a corteza, cerebelo y médula espinal Funciones atención, ánimo, sueño y cognición En SNP actúa en neuronas postganglionares del SNS Médula Adrenalina Hormona de estrés Pons R. The phenotypic spectrum of paediatric neurotransmitter diseases and infantile parkinsonism. J Inherit Metab Dis (2009) 32: 321– 332

Metabolismo de las monoaminas Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33

Metabolismo de BH 4 Además cofactor de: 3 NO sintasas Gliceril-eter monooxigenasa Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25 Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis (2009) 32: 333– 342

Metabolismo de BH 4 Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis (2009) 32: 333– 342
 que")
Defectos del metabolismo de BH 4 Descritos inicialmente en pacientes con hiperfenilalaninemia (HFA) que desarrollaron deterioro neurológico progresivo pese a óptimo control metabólico: HFA maligna � Existen 5 condiciones genéticas distintas que afectan la síntesis o regeneración de BH 4 � Debido a actividad defectuosa de hidroxilasas de fenilalanina, tirosina y triptofano Sólo una relativamente benigna Todas pesquisables por HFA en screening neonatal, excepto � GTPCH AD � Déficit de SR Alteración BH 4 solo en cerebro Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis (2009) 32: 333– 342

Trastorno del metabolismo de las monoaminas: clasificación clínica Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33

Déficit de BH 4 Déficit de GTPCH AR Déficit de PTPS Con HFA Déficit de PCD Errores congénitos del metabolismo de las pterinas Déficit de DHPR Déficit de GTPCH AD Sin HFA Déficit de SR

Clínica: Síntomas y signos sugerentes Manifestaciones extrapiramidales Síntomas autonómicos Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25 Assmann B, et al. Approach to the Diagnosis of Neurotransmitter Diseases Exemplified by the Differential Diagnosis of Childhood-Onset Dystonia. Ann Neurol

Clínica: Síntomas y signos sugerentes Pons R. The phenotypic spectrum of paediatric neurotransmitter diseases and infantile parkinsonism. J Inherit Metab Dis (2009) 32: 321– 332

Clasificación Enfermedades de los NT que se presentan entre los 0 y 2 años Encefalopatía grave de inicio precoz, progresiva, asociada a alteraciones de las monoaminas Deficiencia de Tirosina Hidroxilasa (TH) Deficiencia de 1 aminoácido decarboxilasa (AADC) Déficit de triptófano hidroxilasa (TPH) ECM de las Pterinas • Déficit de Sepiapterina Reductasa (SR) • Deficiencia de GTPCH I (AR) Enfermedades de los NT que se presentan a partir de los 2 años Clínica marcada con trastornos del movimiento • Deficiencia de GTPH autosómica dominante (Segawa- distonía sensible a DOPA)
")
Diagnóstico Historia clínica Examen físico Estudio: � Bioquímico � Ensayo enzimático (para algunos trastornos) � Estudio de mutaciones genéticas Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33

Diagnóstico diferencial Distonías relacionadas con trastornos neurometabólicos Distonías primarias y paroxísticas de la infancia DYT-1 Coreoatetosis kinesogénica paroxística Distonía no kinesogénica Distonía inducida por ejercicio Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Diagnóstico diferencial Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Screening en RN de HFA Blau N et al. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH 4) deficiencies. . Mol Genet Metab. 2011; 104 Suppl: S 2 -9

Estudio Medición en sangre y orina de pterinas y metabolitos de aminas biógenas Concentración NT en LCR � Dado que hay trastornos de la vía de las pterinas que cursan sin hiperfenilalaninemia � Fenilalanina hepática intacta Desafíos técnicos Labilidad de BH 4 en LCR muestra en N líquido o hielo seco Almacenar -80°C Variabilidad en niveles de NT durante el día Alteraciones secundarias de los metabolitos de monoamina en LCR: convulsiones, hipoxia, infecciones o condiciones neurogenéticas correlacionar con la clínica Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33

Estudio Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Estudio Actividad enzimática � No de rutina, se prefiere estudio genético � Puede ser útil para evaluar actividad GTPCH en fibroblastos en pacientes con sospecha de forma AD, particularmente si el perfil de NT en LCR es atípico y el screening GCH 1(-) Carga oral PHE ↑ PHE/tirosina normal Déficit en metabolismo BH 4 Si normaliza con carga PHE+ BH 4 > orientador Test de carga de fenilalanina � Útil en trastornos del metabolismo pterinas SIN HFA � Carga oral de PHE � Falsos positivos y negativos, interpretar con precaución Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33

Diagnóstico Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33

Diagnóstico Friedman et al. Sepiapterin Reductase Deficiency: A Treatable Mimic of Cerebral Palsy. Ann Neurol 2012; 71: 520– 530

A Defecto del metabolismo de las pterinas sin HFA

Defecto del metabolismo de las pterinas sin HFA A. 1 Déficit de GTPCH AD o Enfermedad de Segawa También llamada distonía respondedora a dopa, DYT 5 a o Distonía hereditaria progresiva con marcada fluctuación diurna Descrita por Segawa et al. en 1971: enfermedad hereditaria de los ganglios basales con marcada fluctuación diurna Mutación en el gen de GTP ciclohidrolasa 1 Clínicamente existen 2 tipos: Distonía postural Distonía de acción • Síntomas similares inter e intrafamiliar • Síntomas con variaciones intrafamiliares Depende de la familia o el loci de la mutación Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain & Development 33 (2011) 195– 201 Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Defecto del metabolismo de las pterinas sin HFA Déficit de GTPCH AD o Enfermedad de Segawa: A. 1 Fisiopatología Mutación en el gen de GTP ciclohidrolasa 1 (GCH-1) � Ubicado en cromosoma 14 q 22. 1 -q 22. 2 � Deficiencia parcial de la actividad de la enzima Herencia AD, mayor penetrancia en mujeres (87%) que en hombres (38%) � >100 mutaciones en región codifcante independientes descritas La misma intrafamiliar, distintas interfamiliar Aprox 40% no se determina la mutación Alteraciones en deleción de intrones genómicos, deleción grande del gen, duplicación o inversión intragénica Mutación de un gen regulador que modifique función enzmática aún no determinada Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain & Development 33 (2011) 195– 201

Defecto del metabolismo de las pterinas sin HFA Déficit de GTPCH AD o Enfermedad de Segawa: A. 1 Fisiopatología Defecto afecta la síntesis de BH 4 Debiera afectar triptofano hidroxilasa (TPH) tanto como la TH (vía serotonina igual de afectada que la de dopamina) Mutación heterocigota déficit parcial de BH 4 TH > TPH afinidad por BH 4 TH se afecta selectivamente por su mayor afinidad a BH 4 En condiciones en que BH 4 está marcadamente disminuida afectación de TH=TPH síntomas de vía Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain & Development 33 (2011) 195– 201 serotonina Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Defecto del metabolismo de las pterinas sin HFA Déficit de GTPCH AD o Enfermedad de Segawa: A. 1 Fisiopatología Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain & Development 33 (2011) 195– 201

Defecto del metabolismo de las pterinas sin HFA A. 1 Déficit de GTPCH AD o Enfermedad de Segawa: Clínica Distonía fluctuante Temblor CI normal Espectacular respuesta a L-DOPA En la mayoría de los casos el síntoma inicial es postura distónica de una extremidad inferior, pie equinovaro, alrededor de los 6 años Progresa a todos los miembros y tronco (adolescencia) Rigidez se agrava progresivamente hasta los 20 años La progresión se relentece en la 2° década Se hace estacionaria en los 30 Luego de 10 años aparece temblor postural 8 -10 Hz en EESS, se expande a todos los miembros a los 30 La locomoción se preserva Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain & Development 33 (2011) 195– 201 Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Defecto del metabolismo de las pterinas sin HFA A. 1 Déficit de GTPCH AD o Enfermedad de Segawa: Clínica Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain & Development 33 (2011) 195– 201

Defecto del metabolismo de las pterinas sin HFA A. 1 Déficit de GTPCH AD o Enfermedad de Segawa: Clínica En el tipo Distonía de acción, además aparecen movimientos de una extremidad superior o retrocollis de acción alrededor de los 8 años � Puede asociarse a crisis oculógiras � Tortícolis y calambre del escribidor aparecen en la adultez � En la familia hay casos de inicio en la adultez con calambre del escribidor, tortícolis o rigidez generalizada con temblor postural, sin posturas distónicas ni progresión aparente � Predominancia masculina Inicio en la niñez: Marcada fluctuación diurna, luego se atenúa en la adultez Marcada predominancia femenina (4: 1) Inicio precoz: RDSM Estancamiento del crecimiento longitudinal con el inicio de la distonía Postural 18: 1 Acción 2: 1 Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain & Development 33 (2011) 195– 201

Defecto del metabolismo de las pterinas sin HFA Déficit de GTPCH AD o Enfermedad de Segawa: Examen A. 1 físico Rigidez no plástica � Casos con temblor sin rueda dentada � Asimetría rigidez y temblor En niñez ROT ↑, clonus (+), con plantares flexores Enfermedad avanzada: pulsión, sin freezing Cerebelo y sensibilidad sin alteraciones Inicio precoz: hipotonía tronco, RDSM motor, camptocormia en la niñez tardía, parkisonismo en la adultez Talla baja se recupera si se inicia tratamiento antes de la adolescencia Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain & Development 33 (2011) 195– 201

Defecto del metabolismo de las pterinas sin HFA A. 1 Déficit de GTPCH AD o Enfermedad de Segawa: Estudio RM cerebro: normal Fenilalanina sérica normal � Medición en LCR de NT � Adecuada conversión intrahepática de fenilalanina a tirosina Niveles bajos de ácido homovanílico, neopterina, 3 -Ometildopa (y BH 4) Análisis genético � Rendimiento 50% (aún no se conoce todas las mutaciones) Considerar presentaciones atípicas: similar a diplejia espástica, distonía de ee asimétrica, calambre del escribiente Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain & Development 33 (2011) 195– 201 Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Defecto del metabolismo de las pterinas sin HFA Déficit de GTPCH AD o Enfermedad de Segawa: A. 1 Tratamiento L-Dopa en dosis bajas muestra marcado beneficio � Asilada 20 mg/kg/d o � 4 -5 mg/kg/d asociada a inhibidor de decarboxilasa � 20% pacientes presentan diskinesias: Efectividad mantenida en algunas familias Necesario en algunas familias años después del inicio del tto Movimientos coreicos con ascenso rápido de dosis o inicio con dosis alta Responde con la disminución de dosis o titulación adecuada Respuesta incompleta en distonía de acción y trastornos asociados Retrocolis de acción y crisis oculógiras pueden empeorar inicialmente Más riesgo de diskinesia inducida por L-dopa Anticolinérgicos: efecto marcado sobre distonía, no en temblor BH 4: asociado con L-dopa se ha reportado efecto moderado, sin efecto en monoterapia (Se usa muy raramente) Segawa M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain & Development 33 (2011) 195– 201 Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25|

Defecto del metabolismo de las pterinas sin HFA A. 2 Déficit de Sepiapterina Reductasa (SR) Trastorno de NT sensible a dopa Muy infrecuente Causado por mutación del gen SPR (2 p 14 -p 12) � Hasta el momento 14 mutaciones diagnosticadas � Herencia AR Dill P, et al. Child neurology: paroxysmal stiffening, upward gaze, and hypotonia: hallmarks of sepiapterin reductase deficiency. Neurology. 2012 Jan 31; 78(5): e 29 -32 Arrabal L, et al. Genotype–phenotype correlations in sepiapterin reductase deficiency. A splicing defect accounts for a new phenotypic variant. Neurogenetics

Defecto del metabolismo de las pterinas sin HFA A. 2 Déficit de Sepiapterina Reductasa (SR) Clínica: inicio de síntomas <1 año � � Trastorno motor respondedora a dopa con fluctuaciones diurnas, en la mayoría de los casos asociados con DI y disfunción neurológica severa Tríada Diagnóstico: análisis de LCR metabolitos de aminos biógenas y especies pterinas Retraso en el diagnóstico promedio 9, 1 años Crisis oculógiras Hipertonía paroxística Hipotonía Dill P, et al. Child neurology: paroxysmal stiffening, upward gaze, and hypotonia: hallmarks of sepiapterin reductase deficiency. Neurology. 2012 Jan 31; 78(5): e 29 -32 Arrabal L, et al. Genotype–phenotype correlations in sepiapterin reductase deficiency. A splicing defect accounts for a new phenotypic variant. Neurogenetics 2011, 12: 183– 91

Defecto del metabolismo de las pterinas sin HFA A. 2 Déficit de Sepiapterina Reductasa (SR): clínica Dill P, et al. Child neurology: paroxysmal stiffening, upward gaze, and hypotonia: hallmarks of sepiapterin reductase deficiency. Neurology. 2012 Jan

Defecto del metabolismo de las pterinas sin HFA A. 2 Déficit de SR: Clínica >65% 4565% Relacionados con alteración vía de dopamina y serotonina Manifestaciones cardinales: distonía y fluctuación diurna no son universales (79%) A menudo ausentes precozmente Distonía sólo 50% pctes con dx >4 años Friedman et al. Sepiapterin Reductase Deficiency: A Treatable Mimic of Cerebral Palsy. Ann Neurol 2012; 71: 520– 530

Defecto del metabolismo de las pterinas sin HFA A. 2 Déficit de SR: Clínica Relacionados con alteración vía de dopamina y serotonina Microcefalia Síntomas pueden interrumpirse por movimientos voluntarios DSM normal: mutaciones que conservan función parcial Síntomas particulares son edad específicos Dill P, et al. Child neurology: paroxysmal stiffening, upward gaze, and hypotonia: hallmarks of sepiapterin reductase deficiency. Neurology. 2012 Jan 31; 78(5): e 29 -32

Defecto del metabolismo de las pterinas sin HFA A. 2 Déficit de SR: Diagnóstico Fenilalanina sérica normal, pterinas normales en orina Estudio de LCR � � ↑ pterinas (↑ ↑sepiapterina) ↓ ácido 5 hidroxindolacetico y ácido homovanílico Actividad sepiapterina reductasa en fibroblastos: confirma Análisis mutación Dill P, et al. Child neurology: paroxysmal stiffening, upward gaze, and hypotonia: hallmarks of sepiapterin reductase deficiency. Neurology. 2012 Jan 31; 78(5): e 29 -32

Defecto del metabolismo de las pterinas sin HFA A. 2 Déficit de SR: Tratamiento Sustitución de los precursores Ldopa y 5 -hidroxitriptifano L-dopa/carbidopa o benserazida (5 mg/kg/d) + 5 -hidroxitriptofano (2, 5 mg/kg/día) L-dopa/carbidopa (2 mg/kg/día) � Frecuencia 2 -5 veces al día Raro: + selegilina o sertralina Mejoría clínica rápida (en horas) del déficit motor Parcial o total Sin efecto sobre el rendimiento cognitivo DI leve asevera CI normal 3/21 pacientes Vómitos severos en algunos pacientes con 5 -hidroxitriptofano Dill P, et al. Child neurology: paroxysmal stiffening, upward gaze, and hypotonia: hallmarks of sepiapterin reductase deficiency. Neurology. 2012 Jan 31; 78(5): e 29 -32

B Defecto del metabolismo de las pterinas con HFA

Defecto del metabolismo de las pterinas con HFA Defecto del metabolismo de las pterinas con B HFA

Defecto del metabolismo de las pterinas con HFA B. 1 Déficit de GTPCH autosómica recesiva Mutación rara de GTPCH (solo 5 de 104 alelos mutantes) � Causa reducción severa de BH 4 hiperfenilalaninemia (HFA) � Distonía respondedora a dopa de herencia AR generalmente por déficit de otras enzimas, sin HFA � Fenotipo más complejo, respuesta moderada a L-dopa (síndromes DRD plus) � Manifestación cardinal HFA � Casos reportados de Sd. DRD-plus de GCH 1 AR sin HFA Clínica � <10% casos déficit GTPCH 1 Inicio en la infancia, generalmente con RDSM, piramidalismo, distonía, temblor, convulsiones y disfunción autonómica Diagnóstico diferencial: otros trastornos de NT, trastornos metabólicos, PC Thony B, Blau N. Mutations in the BH 4 -Metabolizing Genes GTP Cyclohydrolase I, 6 -Pyruvoyl-Tetrahydropterin Synthase, Sepiapterin Reductase, Carbinolamine-4 a- Dehydratase, and Dihydropteridine Reductase. Hum Mutat 27(9), 870– 878, 2006 Sato H et al. Early replacement therapy in a first Japanese case with autosomal recessive GTPCH 1 deficiency with a novel point mutation. Brain Dev. 2013 May 6. pii: S 0387 -Bruggemann N et al. Beneficial Prenatal Levodopa Therapy in Autosomal Recessive Guanosine Triphosphate Cyclohydrolase 1 Deficiency. Arch Neurol. 2012; 69(8): 1071 -1075
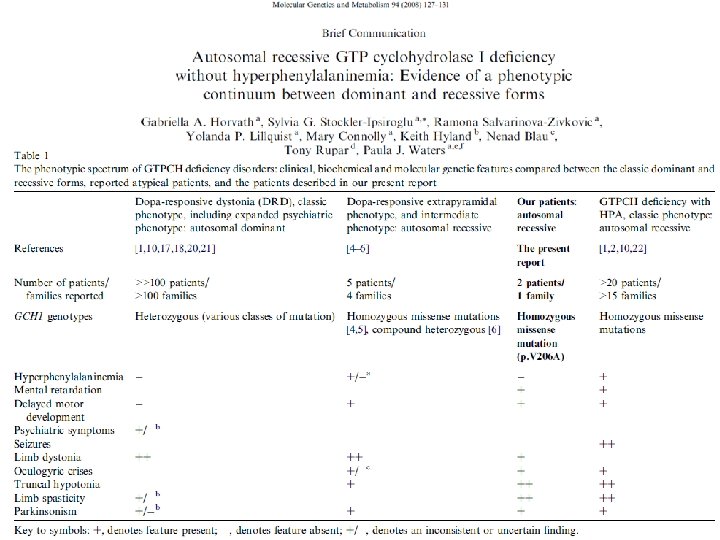

Defecto del metabolismo de las pterinas con HFA B. 1 Déficit de GTPCH autosómica recesiva Diagnóstico La mayoría en screening neonatal por HFA Tratamiento Suplementación BH 4 � Dosis 1 -10 mg/kg/d aumentan la actividad de la fenialanina hidroxilasa hepática normalización niveles fenilalanina � Cantidad que ingresa al cerebro es insuficiente para síntesis de NT ↓ biopterina y neopterina en orina LCR: ↓ HVA, 5 -HIAA y pterinas Medición de actividad de enzima puede ser útil para confirmar el diagnóstico Identificación de mutación en GCH 1 homocigota o heterocigota compuesta L-dopa y 5 -hidroxitriptofano Inhibidores de monoamino oxidasa Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 Bruggemann N et al. Beneficial Prenatal Levodopa Therapy in Autosomal Recessive Guanosine Triphosphate Cyclohydrolase 1 Deficiency. Arch Neurol.

Defecto del metabolismo de las pterinas con HFA Déficit de 6 -piruvoil-tetrahidropterina sintasa B. 2 (PTPS) Trastorno más frecuente del metabolismo de BH 4 Forma más prevalente y heterogénea de HFA no atribuida a deficiencia de fenilalanina oxidasa Frecuente pesquisa en screening neonatal Fenotipo PKU+ manifestaciones neurológicas de déficit de monoaminas Mutación gen PTS (cr 11 q 22. 3 q 23. 3) 2 presentaciones: forma típica/severa vs atípica/periférica Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Defecto del metabolismo de las pterinas con HFA B. 2 Déficit de PTPS: fisiopatología Mutación gen PTS (cr 11 q 22. 3 q 23. 3) � 6 exones � >50 mutaciones descritas, alta heterogeneidad alélica � Aparente buena relación genotipo -fenotipo � 2/3 asociadas con forma severa � Fenotipo severo: cambio de marco de lectura o alteración de unión de proteína a zinc o de oligomerización Pérdida de actividad PTPS disminuye sustancialmente niveles BH 4 alteración producción de dopamina y Thony B, Blau N. Mutations in the BH 4 -Metabolizing Genes GTP Cyclohydrolase I, 6 -Pyruvoyl-Tetrahydropterin Synthase, Sepiapterin Reductase, serotonina Carbinolamine-4 a- Dehydratase, and Dihydropteridine Reductase. Hum Mutat 27(9), 870– 878, 2006 Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Defecto del metabolismo de las pterinas con HFA B. 2 Déficit de PTPS: Clínica Forma leve (20%) Forma severa (80%) • Curso neurológico normal • Pronóstico excelente LCR con HVA y 5 -HIAA normal • Compromiso neurológico • RNPT y BPN LCR con HVA y 5 HIAA bajo Inicio en periodo lactante Inicial hipotonía axial, luego hipertonía apendicular, bradikinesia, rigidez en rueda dentada, distonía generalizada y marcada fluctuación diurna � También dificultades en deglución, crisis oculógiras, somnolencia, irritabilidad, hipertermia y crisis generalizada, coreoatetosis, hipersalivación, rash con eczema y muerte súbita � Síntomas neuropsiquiátricos Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 Pearl PL. Monoamine neurotransmitter deficiencies. Handb Clin Neurol. 2013; 113: 1819 -25

Defecto del metabolismo de las pterinas con HFA B. 2 Déficit de PTPS: Estudio HFA en test gota de sangre frecuente En orina ↓ biopterina, ↑ neopterina Puede haber ↑ prolactina en sangre LCR � ↑ neopterina � ↓ otros metabolitos biopterinas, HVA y 5 -HIAA Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 Niu D-M. Disorders of BH 4 metabolism and the treatment of patients with 6 -pyruvoyl-tetrahydropterin synthase deficiency in Taiwan. Brain Dev. 2011; 33(10): 847 -55

Defecto del metabolismo de las pterinas con HFA B. 2 Déficit de PTPS: Tratamiento Administración BH 4 � Normaliza niveles de fenilalanina � Iniciar con 2 mg/kg/d y ajustar para mantener [PHE] <120 u. M � No cruza BHE Asociado a L-DOPA y 5 hidroxitriptofano � Dosis óptima difícil de determinar � Precaución con efectos secundarios Outcome Serie Taiwanesa (n=12) Serie italiana (n=19) L-dopa inicio con 2 mg/kg/d ↑ 1 mg c/25 días Target 1015 mg/kg/d 5 Hidroxitriptofano inicio 1 mg/kg/d ↑ 1 mg c/2 -5 días Target 5 mg/kg/d • CI 96. 7 (± 9. 7; rango: 86– 119) • Forma leve (n=6): DSM normal • Forma severa (n=13) 12 compromiso neurológico grave, DSM normal 4/13 Forma severa de la enfermedad: outcome mejora si tratamiento se inicia precozmente Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 Niu D-M. Disorders of BH 4 metabolism and the treatment of patients with 6 -pyruvoyl-tetrahydropterin synthase deficiency in Taiwan. Brain Dev. 2011; 33(10): 847 -55

Defecto del metabolismo de las pterinas con HFA B. 3 Déficit de dihidropteridina reductasa (DHPR) Defecto en la regeneración de BH 4 luego de la hidroxilación de sustratos y la acción de la carbinolamina dehidratasa ↓ BH 4 ↓ serotonina y dopamina Usualmente detectada en screening neonatal por HFA Clínicamente es más severa que otros trastornos del metabolismo de las pterinas � RDSM pese a tratamiento Causado por mutación en gen QDPR (4 p 15. 31) � � >34 mutaciones descritas Buena relación genotipo-fenotipo Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis (2009) 32: 333– 342 Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 Thony B, Blau N. Mutations in the BH 4 -Metabolizing Genes GTP Cyclohydrolase I, 6 -Pyruvoyl-Tetrahydropterin Synthase, Sepiapterin Reductase, Carbinolamine-4 a- Dehydratase, and Dihydropteridine Reductase. Hum Mutat 27(9), 870– 878, 2006

Defecto del metabolismo de las pterinas con HFA B. 3 Déficit de DHPR: Clínica y diagnóstico Inicio en periodo RN o infancia precoz: dificultades para alimentarse, disfunción bulbar, hipersalivación, microcefalia Evolucionan con RDSM, hipertonía tronco y miembros, diskinesias, temblor, distonía, coreoatetosis y convulsiones Mayor riesgo de muerte súbita Prueba de la gota de sangre RN: HPA � ↓ Actividad DHPR marcadamente LCR: ↓ HVA, 5 -HIAA y folato, ↑biopterina. No siempre déficit BH 4 RNM: se puede ver alteraciones SB y calcificaciones de ganglios basales � Reversibles con suplementación de ácido folínico DHPR mantiene folato en su forma activa, ↑q-BH 2 ↓folato en cerebro Deterioro neurológico también atribuible a ↓ folato en cerebro Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis (2009) 32: 333– 342 Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33

Defecto del metabolismo de las pterinas con HFA B. 3 Déficit de DHPR: Tratamiento Suplementación BH 4 Restricción PHE en la dieta Precursores de monoaminas (l-dopa y 5 hidroxitriptofano) IMAOs (selegilina, etc) Suplementar ácido folínico Se ha registrado buen pronóstico con tratamiento precoz Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis (2009) 32: 333– 342 Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33

Defecto del metabolismo de las pterinas con HFA Déficit de Pterin-4α-carbinolamina dehidratasa B. 4 (PCD) Se requiere para la regeneración de BH 4 Mutación gen PCDB 10 q 22 � 9 mutaciones descritas En RN HPA leve con ↑ 7 biopterina persistentemente en orina � PHE se normaliza Se ha reportado hipotonía neonatal transitoria, la mayoría asintomática La mayoría no desarrolla síntomas o signos neurológicos, no se detecta alteraciones NT Pronóstico excelente en Longo N. Disorders of biopterin metabolism. J Inherit Metab Dis (2009) 32: 333– 342 general Kurian MA, et al. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol 2011; 10: 721– 33 Thony B, Blau N. Mutations in the BH 4 -Metabolizing Genes GTP Cyclohydrolase I, 6 -Pyruvoyl-Tetrahydropterin Synthase, Sepiapterin Reductase,

Resumiendo Campeau PM, et al. Neurotransmitter diseases and related conditions. Mol Genet Metab. 2007 Nov; 92(3): 189 -97

Resumiendo En nuestra paciente: antecedente familiar de trastorno de la marcha en 3 generaciones, en familiares de primera línea. Inversión y rotación interna de EEII, con fluctuaciones diurnas que inició a los 9 años Campeau PM, et al. Neurotransmitter diseases and related conditions. Mol Genet Metab. 2007 Nov; 92(3): 189 -97

Conclusiones Grupo de trastornos neurológicos en expansión � Muchos se presentan en periodo lactante o infancia precoz Importante reconocerlos porque frecuentemente se diagnostican erroneamente y muchos tienen buena respuesta clínica a tratamiento Descripción de espectro fenotípico ha mejorado por mayor conciencia de los clínicos, tests bioquímicos más confiables, tests genético moleculares han mejorado
- Slides: 63