Thrombotic Thrombocytopenic Purpura History In 1924 Dr Eli

Thrombotic Thrombocytopenic Purpura

History • In 1924, Dr. Eli Moschcowitz described a 16 - year old girl with abrupt onset of petechiae, pallor, followed by paralysis, coma, and death. • Autopsy showed ‘hyaline’ thrombi occluding terminal arterioles and capillaries. British J of Hematology 2000

Definition • Syndrome of Coomb’s negative microangiopathic hemolysis and thrombocytopenia in the absence of an alternative explanation for these manifestations. • Presence of Fever, Neurological and renal abnormalities : classic Pentad. Rock GA, Br J Hematology 2000

Clinical Presentation • • • Approximately 1000 new cases occur each year Common in middle aged group, median age-40 Female: male (2: 1). Acute onset and fulminant course Mortality rate >90% in pre-pheresis era. Relapse rates, 10 -40% ranging from months to years have been reported. Shumak KH, Ann Intern Med 1995

Clinical Features • Renal abnormalities – Proteinuria/hematuria > oliguria/ARF • Neurological abnormalities – Mental status changes> focal abnormalities. • • Profound weakness related to anemia. Abdominal pain, nausea, vomiting & diarrhea Fever without chills. Primary/idiopathic TTP vs. secondary

Secondary TTP • Drug-induced – Acute immune mediated: Ticlopidine & plavix. – Dose-related: mitomycin, tacrolimus, pencillin, cyclosporine, cisplatin, bleomycin, OCP – Quinine: HUS like illness. • • Pregnancy and post-partum. Allogenic bone marrow transplant. Autoimmune disorders (SLE, scleroderma) HIV infection. George, Blood Aug 2000

Pathogenesis • Deficiency of VWF-cleaving protease – Termed ADAMTS 13 ( “a disintegrin-like and metalloprotease with thrombospondin type I repeats – Corresponding gene : chromosome 9 q 34. – Familial recurrent TTP: constitutional deficiency – Acquired/Idiopathic : transient auto-antibodies – HUS : normal levels of enzyme
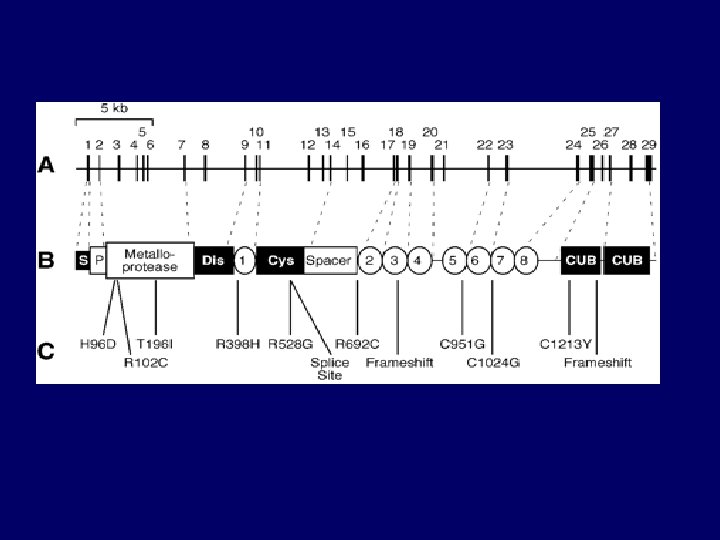

ULVWF Model
 – Microangiopathic")
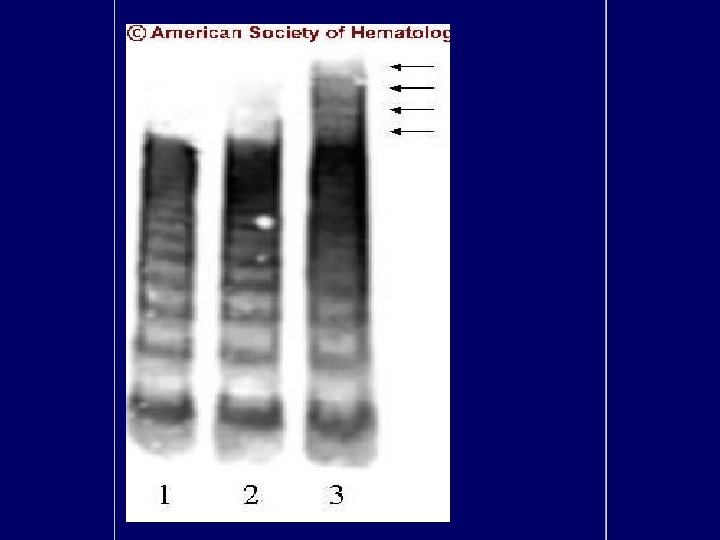
Diagnosis • Primary diagnostic criteria – Thrombocytopenia ( often below <20, 000) – Microangiopathic hemolytic anemia • Negative Coomb’s test. • Fragmented red cells (schistocytes) on peripheral smear • LDH elevation is the hallmark of RBC destruction and tissue injury related to ischemia. • Presence of above criteria is sufficient to establish presumptive diagnosis & begin PE George, Blood Aug 2000
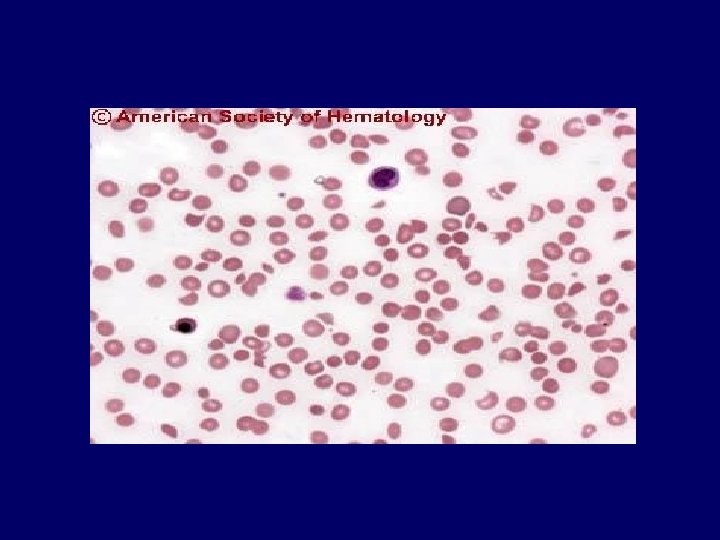
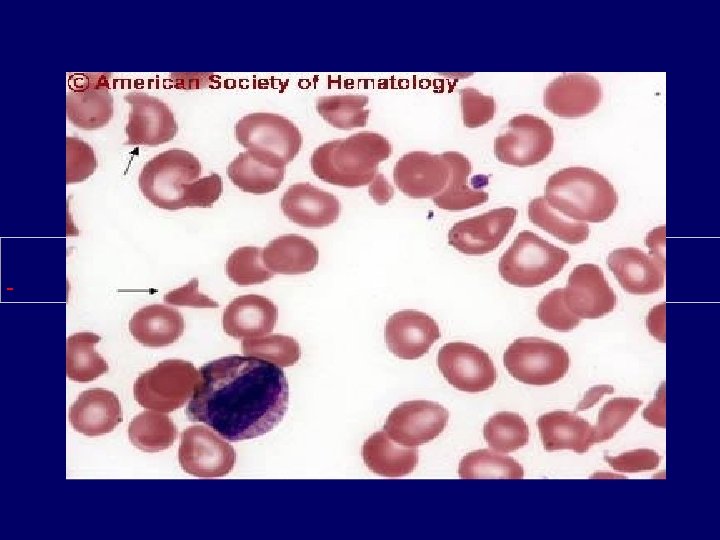
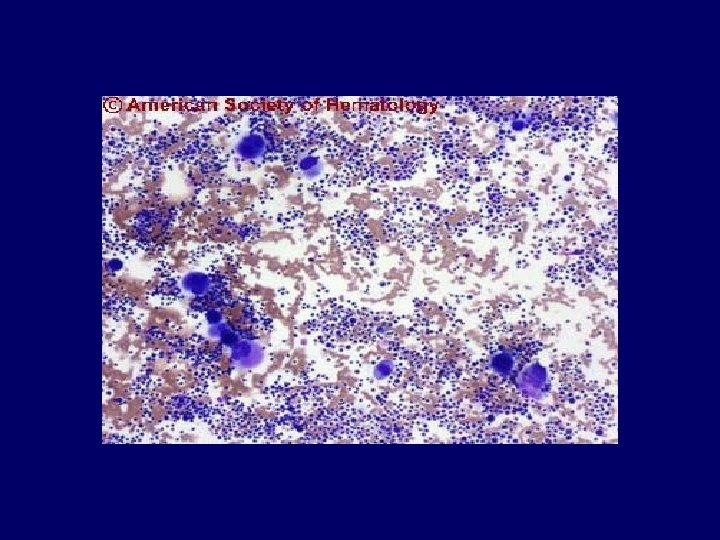
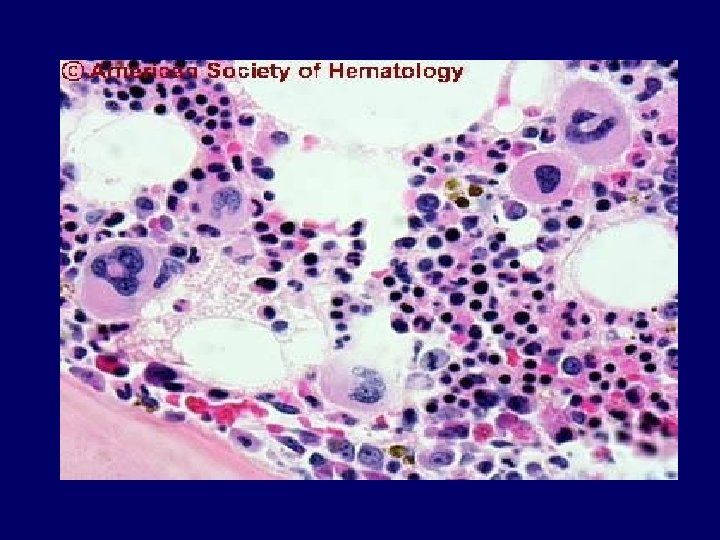

Diagnosis • At present there are no confirmatory test. • Other features in pentad support the diagnosis. • Tests for ADAMTS 13 deficiency or inhibitors are not readily available and lack standardization.

Differential Diagnosis • Disseminated intravascular coagulation. • Sepsis: cytomegalovirus, rocky mountain spotted fever, meningococcemia. • Preeclampsia/eclampsia, HELLP. • Disseminated malignancy. • Hemolytic-uremic syndrome • Evans syndrome • Malignant hypertension.

Treatment • Plasma exchange: – Untreated TTP has 80 -90% mortality. – Removes ULv. WF multimers, autoantibody and replaces metalloproteinase. – Randomized controlled trial (Rock et al, 1991) – FFP as the replacement fluid is most widely used and cost effective.
 – Theoretically superior to FFP in")
Treatment • Cryosupernatant plasma (Rock et al 2000) – Theoretically superior to FFP in refractory disease – Removal of cryoprecipitate from donor plasma results in removal of v. WF ( only 18%), with no change in metalloproteinase concentration. • Solvent-detergent plasma (Moake et al 1998) – Lacks high molecular weight forms of VWF – Inactivates lipid-enveloped viruses. – Drawback: parvovirus & hep A not inactivated.

Response To Treatment • MS changes improve dramatically. • Thrombocytopenia require several days. • Parameters of hemolysis improve promptly, yet anemia may continue to worsen. • Recovery from renal failure is unpredictable and often slow. • Prolonged courses of PE, with frequent exacerbations is characteristic of idiopathic TTP

Complications of Plasma Exchange • Central venous catheter-related Insertion procedure • 4% Sepsis • 15% Thrombosis • 10% • Plasma-related Allergic • 4% Infection • 0 • Instrument-related Unintentional plateletpheresis American Society of Hematology 2002

Duration of treatment. • No studies precisely determine optimal schedule • AABB extracorporeal therapy committee: daily PE until plt ct > 150 k for 2 -3 days. • American Society for Apheresis: daily PE until Plt > 100 k, complete normalization of LDH. • Tapering schedule to 3 times per week after sustained response is highly recommended.

Treatment • Avoid prophylactic platelet transfusion (Gordon et al , 1987; Harkness et al 1981) – Unless life-threatening bleeding is present. – Provide additional substrate for thrombus formation. – MI and strokes have reportedly occurred after transfusion. Conn's current therapy ; 2004
, dipyrimadole ( 400 mg) –")
Adjuvant Therapy • Antiplatelet agents: – Aspirin (325 mg), dipyrimadole ( 400 mg) – Ticlopidine maintenance for 1 year. • Corticosteroids – Presence of auto antibodies to ADAMTS 13 supports the autoimmune disease. – Reserved for patients refractory to PE. British J of Hemat 2000
 • Chemotherapy: Cytoxan, Vincristine, Rituxan, CHOP. •")
Treatment • Splenectomy (Crowther et al, 1996) • Chemotherapy: Cytoxan, Vincristine, Rituxan, CHOP. • High- dose IV Ig. G • Protein A immunoadsorption columns. British J of Hematology 200
- Slides: 25