Thalassemia Thalassemia Thalassemia is an inherited blood disorder

Thalassemia

Thalassemia: • Thalassemia is an inherited blood disorder in which the body makes an abnormal form of hemoglobin. Hemoglobin is the protein molecule in red blood cells that carries oxygen. Hemoglobin is formed by the combination of heme with globin (protein). Globin is made up of four polypeptide chains (an oligomeric protein). Two of these polypeptides are known as alpha (α) and the other two are known as beta (β). • The disorder results in excessive destruction of red blood cells, which leads to anemia. Anemia is a condition in which body doesn’t have enough normal, healthy red blood cells. • Thalassemia is inherited, meaning that at least one of parents must be a carrier of the disease. It’s caused by either a genetic mutation or a deletion of certain key gene fragments
:")
Types of thalassemia: • There are three main types of thalassemia (and four subtypes): 1. Beta thalassemia, which includes the subtypes major and intermedia 2. Alpha thalassemia, which include the subtypes hemoglobin H and hydrops fetalis 3. Thalassemia minor All of these types and subtypes vary in symptoms and severity. The onset may also vary slightly.

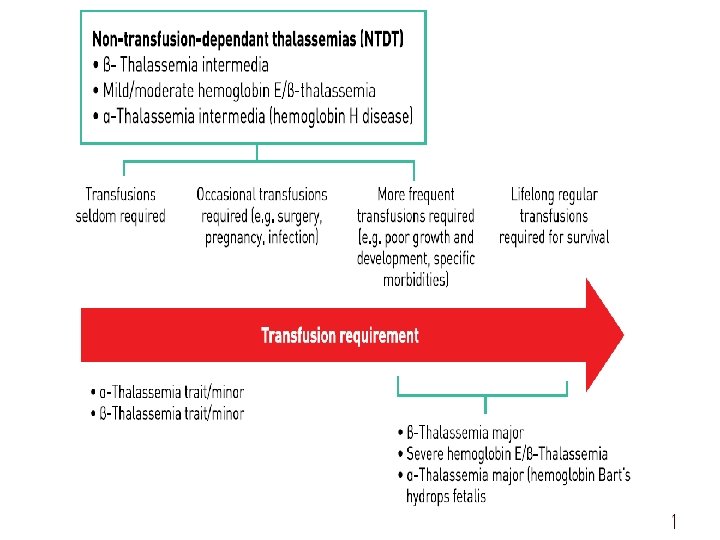
Beta thalassemia: • Beta thalassemia occurs when the body can’t produce beta globin. Two genes, one from each parent, are inherited to make beta globin. This type of thalassemia comes in two serious subtypes: thalassemia major (Cooley’s anemia) and thalassemia intermedia. A. Thalassemia major: is the most severe form of beta thalassemia. It develops when beta globins genes are missing. The symptoms of thalassemia major generally appear before a child’s second birthday. The severe anemia related to this condition can be life-threatening. Other signs and symptoms include:

fussiness paleness frequent infections a poor appetite failure to thrive jaundice, which is a yellowing of the skin or the whites of the eyes enlarged organs This form of thalassemia is usually so severe that it requires regular blood transfusions. B. Thalassemia intermedia: is a less severe form. It develops because of alterations in both beta globin genes. People with thalassemia intermedia don’t need blood transfusions.

Alpha thalassemia occurs when the body can’t make alpha globin. In order to make alpha globin, you need to have four genes, two from each parent. This type of thalassemia also has two serious types: hemoglobin H disease and hydrops fetalis. Hemoglobin H : develops as when a person is missing three alpha globin genes or experiences changes in these genes. This disease can lead to bone issues. The cheeks, forehead, and jaw may all overgrow.

Additionally, hemoglobin H disease can cause: jaundice an extremely enlarged spleen malnourishment Hydrops fetalis : is an extremely severe form of thalassemia that occurs before birth. Most individuals with this condition are either stillborn or die shortly after being born. This condition develops when all four alpha globin genes are altered or missing.

Thalassemia minor: People with thalassemia minor don’t usually have any symptoms. If they do, it’s likely to be minor anemia. The condition is classified as either alpha or beta thalassemia minor : In alpha minor cases, two genes are missing. In beta minor, one gene is missing. The lack of visible symptoms can make thalassemia minor difficult to detect. It’s important to get tested if one of your parents or a relative has some form of the disease

Diagnosis: 1. Lab test: blood tested for anemia and abnormal hemoglobin. A lab technician will also look at the blood under a microscope to see if the red blood cells are oddly shaped, pale, with low level , smaller than expected or with uneven distribution of HB(bull eye appearance). Abnormally shaped red blood cells are a sign of thalassemia. 2. hemoglobin electrophoresis: This test separates out the different molecules in the red blood cells, allowing them to identify the abnormal type. 3. physical examination: might also help your doctor make a diagnosis. For example, a severely enlarged spleen might suggest that you have hemoglobin H disease

Treatment: • The treatment for thalassemia depends on the type and severity of disease involved. Select acourse of treatment that will work best for a particular case. Some of the treatments include: • blood transfusions • bone marrow transplant • medications and supplements • possible surgery to remove the spleen or gallbladder Prognosis: The outlook depends on the type of the disease. People who have mild or minor forms of thalassemia can typically lead normal lives. In severe cases, heart failure is a possibility

Blood Transfusion: • How often you need to have transfusions depends on the type of thalassaemia you have. • regular blood transfusions given every three to four weeks (start when Hb level <7 g/dl). • Transfusions correct the anaemia, enable growth and normal activity levels, prevent enlargement of the spleen and inhibit the erythroid marrow expansion. • The most important long-term problem associated with regular transfusions in thalassemia is iron overload. Blood contains iron which cannot be excreted from the body and can be potentially fatal, • SO Patient should not take vitamins or supplements containing iron. If receiving blood transfusion.

a blood transfusion, also need chelation therapy. This generally involves receiving an injection of a chemical that binds with iron and other heavy metals. This help remove extra iron from the body. Iron chelation therapy should be initiated after 10 - 12 transfusions (>20 red cell units) and/or when the serum ferritin level is consistently greater than 1000 μg/L The established regime requires subcutaneous infusions of the chelating agent desferrioxamine given 5 -7 nights per week over 8 -12 hours or oral iron chelator , deferiprone or deferasirox These can help greatly where adherence to parenteral desferrioxamine infusions has been a problem,

The following are the options for blood used for transfusion : Neocytes are young red cells, which are harvested from normal donors. In theory, these red blood cells can prolong patients’ life span and they subsequently need less frequent transfusion. Leukocyte reduced packed RBC These are packed red cells with the level of leukocytes reduced to less than 5 x 106. Supertransfusion regime Under this regime, the pre-transfusion Hb is kept above 12 g/L Hypertransfusion regime The pre-transfusion Hb in this regime is usually above 10 -12 g/L Moderate transfusion regime In the moderate transfusion regime, the pre-transfusion Hb is maintained between 9 -10 g/L.

In some TI children, despite their having Hb levels > 7 g/d. L, growth failure or cosmetic facial and bony abnormalities occur, which may not be reversible unless regular transfusions are started before the age of 6 or 7 years. In older patients, massive splenomegaly is often associated with hypersplenism, which contributes to progressive anemia neutropenia and thrombocytopenia, and it warrants a trial of regular transfusions to improve splenic size and function, although splenectomy may be required. TI patients who develop progressive anemia, fatigue, and cardiopulmonary complications also require regular transfusions to maintain Hb levels > 9 -10 g/d. L

Stem cell or bone marrow transplants • SCT are the only cure for thalassemia, but they're not done very often because of the significant risks involved. • For a stem cell transplant, stem cells from a healthy donor are given through a drip into a vein. These cells then start to produce healthy red blood cells to replace the cells affected by thalassemia. • The main risk of SCT is graft versus host disease, which is a life-threatening problem where the transplanted cells start to attack the other cells in the body. • For people with serious types of thalassemia, the longterm benefits of a stem cell transplant will need to be considered against the possible risks to help determine whether the treatment is suitable.

surgery Splenectomy • The severe hemolysis results in progressive overactivity of the spleen, which eventually aggravates the severity of the anemia and consequently increases transfusion requirements. • After the initiation of a regular transfusion program from an early age, splenomegaly may be averted, . • The therapeutic rationale for splenectomy, particularly in patients with growth retardation and poor health, is to protect against the development of extramedullary hematopoiesis by improving the Hb level, decreasing the transfusion requirement, and consequently reducing iron overload (IO). It should be noted there are patients who are on regular transfusion programs who develop hypersplenism without splenomegaly.

Therefore, we recommend splenectomy when the calculated annual transfusion requirement is > 200 to 220 m. L RBCs/kg per year with a hematocrit of 70% The susceptibility to overwhelming infections after splenectomy can be reduced by immunization with pneumococcal and meningococcal vaccines before splenectomy and antimicrobial prophylaxis with penicillin after splenectomy

Treating other problems Thalassemia can also cause a number of other problems that may need to be managed. For example: • hormone medication may be used to help trigger puberty in children with delayed puberty and to treat low hormone levels • vaccinations and antibiotics may be recommended to prevent and treat infections • thyroid hormones may be used if there's a problem with your thyroid gland (hypothyroidism) • medications called bisphonates may be used to help strengthen the bones if you have fragile bones (osteoporosis) • gallstones may be treated with gallbladder

Nutrition, vitamin support and psychology therapy are other modalities used in the treatment of Thalassemia patients. Vitamin C is only given to those with established depletion and those who are on chelation therapy. For Thalassemic children requiring splenectomy, additional immunization with Haemophilus influenza B, pneumococcal and meningococcal vaccines is usually carried out.

THANK YOU
- Slides: 21