Termodinamica dequilibrio l l Definizione La termodinamica una

Termodinamica d’equilibrio l l Definizione: La termodinamica è una scienza che riguarda i trasferimenti di calore e lo svolgimento di lavoro nel corso di processi chimici e fisici u La termodinamica comporta la predizione dello stato finale di un sistema e del percorso (processo) effettuato per raggiungerlo.

Storia della Termodinamica l l l J. Willard Gibbs analizzò le prime applicazioni di questa scienza alla chimica nel corso di una serie di pubblicazioni (1875 -1878); Questi studi furono svolti sulla base dei primi risultati sulla relazione tra calore e lavoro (ad es. Le macchine a vapore ed il ciclo di Carnot); Tali studi hanno fornito ai geochimici un utile strumento quantitativo per capire e operare previsioni sul comportamento dei sistemi geochimici

Termodinamica: principi generali l un sistema: Una porzione dell’universo che si vuole studiare l Il circostante/l’intorno: La parte adiacente dell’universo prossima al sistema I cambiamenti in un sistema sono associati al trasferimento di energia I sistemi naturali tendono verso stati di minima energia

Sistema isolato – nessun scambio di materia ed energia con l’ambiente esterno chiuso – può scambiare energia ma non materia aperto – può scambiare sia materia che energia

Termodinamica: principi generali Una Fase: una porzione meccanicamente separabile di un sistema: Minerale F Liquido F Vapore Una Reazione: alcune variazioni nella natura o nel tipo di fasi di un sistema: F Le reazioni sono scritte nella forma: reagenti = prodotti

Termodinamica: principi generali La termodinamica studia le proprietà dei sistemi fisici da un punto di vista macroscopico. In termodinamica, scienza nata con l’invenzione delle macchine a vapore e la rivoluzione industriale, si introducono grandezza macroscopiche misurabili, dette grandezze termodinamiche, e si analizzano le relazioni funzionali tra queste grandezze. Il sistema fisico di riferimento della termodinamica è il gas, le cui grandezze termodinamiche sono il volume V, la pressione P e la temperatura T. Ad esempio, nel caso di un gas diluito all’equilibrio termodinamico, gli esperimenti mostrano che vale la seguente equazione di stato PV=n. RT, dove n è il numero di moli ed R=8, 316 J/(mole °K) è la costante dei gas.

Termodinamica: principi generali Processi Isotermico – avviene a T costante Isopiezo – avviene a P costante Isocoro – avviene a V costante Adiabatico – avviene senza guadagno o perdita di calore Lavoro Il lavoro, W, è positivo se esso compare nell’ambinete esterno, negativo se scompare dall’ambiente esterno; Lavoro meccanico = forza x distanza Lavoro termodinamico = pressione x volume PDV

Termodinamica l l l Nulla si crea e nulla si distrugge Quindi, l’energia totale dell’UNIVERSO è costante. L’energia può tuttavia essere convertita da una forma ad un’altra o trasferita da un sistema all’ambiente circostante e viceversa.

Processi spontanei l l I processi spontanei sono quelli che procedono senza un intervento esterno. Il gas nel contenitore B spontaneamente si espanderà nel contenitore A all’apertura del rubinetto, ma l’inverso non avverrà

Processi spontanei I processi che sono spontanei in una direzione non lo sono nella direzione inversa.

Processi spontanei l l l Processi che sono spontanei ad una certa temperatura possono essere non spontanei ad altre temperature. Sopra 0 C il ghiaccio fonde e si scioglie. Sotto 0 C il processo inverso è spontaneo

Ci interesseremo essenzialmente di reazioni chimiche cercando di determinare i criteri di spontaneità cioè di capire in quale direzione esse avvengono spontaneamente.

Processi reversibili In un processo reversibile il sistema cambia in modo tale per cui il sistema e l’ambiente circostante possono tornare al loro rispettivo stato generale.

Processi irreversibili l l l I processi irreversibili non permettono un ritorno alle condizioni prima della variazione. Tutti i processi spontanei sono irreversibili. Tutti i processi reali sono irreversibili.

Principio “ 0” della termodinamica Legge dell’equilibrio termico “Se due oggetti sono in equilibrio termico con un terzo oggetto, allora essi saranno in equilibrio termico fra loro” C A B Se C è il termometro, allora è evidente che non sarà più necessario confrontare tra loro direttamente due sistemi per stabilire il loro mutuo stato termico.

Termodinamica: le prime due leggi l I° Principio: l’energia dell’universo è costante e non varia a causa di trasformazioni fisiche o chimiche di sistemi. Ogni sistema, in un determinato stato, ha una energia interna, che è funzione di stato. Quando un sistema subisce un cambio di stato, la variazione di energia dipende solo dal punto iniziale e finale e non dal percorso del processo DU = (variazione d’energia) Q (calore trasferito) - W (lavoro) Si definisce con entalpia H la grandezza che a V costante (senza che venga effettuato lavoro, dal momento che W=PDV) risulta: DH = Q DH può essere misurato attraverso le variazioni di T (calorimetro), ed è fornita in tabulati per molte sostanze.

Prima legge della termodinamica Legge sulla conservazione dell’energia “Una variazione in energia interna equivale a calore meno lavoro” • Empiricamente si osserva che per ogni percorso che porta un sistema chiuso da uno stato iniziale (P 1, V 1, T 1) ad un nuovo stato (P 2, V 2, T 2), e poi indietro (P 1, V 1, T 1). La somma del calore e del lavoro trasferiti attraverso i “confini” del sistema è 0. • Nè calore nè lavoro sono variabili di stato; le quantità scambiate attorno a percorsi chiusi di calore e lavoro possono essere ≠ 0; solo la somma è conservata. – Quindi, è inappropriato parlare di quantità di lavoro e di calore in un sistema; queste quantità sono usate solamente per “trasferimenti”. • Possiamo però definire una variabile di stato E (energia interna)la cui variazione per un sistema chiuso è
 esprime la quantità di energia che un sistema termodinamico può scambiare")
L' entalpia (H) esprime la quantità di energia che un sistema termodinamico può scambiare con l'ambiente. L'entalpia è definita dalla somma dell'energia interna e del prodotto tra volume e pressione di un sistema. Dalla definizione di entalpia: H = U + p. V dove U è l' energia interna del sistema, p la sua pressione e V il suo volume. U è l'energia totale posseduta da un sistema materiale, intesa come somma dei contributi di energia traslazionale, rotazionale e vibrazionale delle molecole che lo compongono, più il contributo dell'energia dovuto agli elettroni e dell'energia al punto zero (energia fondamentale posseduta a 0 K).
 ma in")
Un tempo si pensava che le reazioni spontanee dovessero essere esotermiche (DH<0) ma in realtà si possono osservare come spontanee diverse reazioni endotermiche (DH>0). Ad esempio sono spontanee reazioni endotermiche quali H 2 O(s) H 2 O(l) Na. Cl(s) Na+(aq) + Cl-(aq) DHfus = +6. 0 k. J/mol DHsol = +6. 4 k. J/mol o termoneutre quali il mescolamento di due gas La spontaneità di una reazione (o in generale di un processo complesso) non è determinata univocamente dalla variazione di energia (o entalpia) ma richiede una nuova grandezza nota come entropia

Termodinamica: le prime 2 leggi l II° Principio: ogni trasformazione spontanea avviene in modo da far aumentare lo stato di disordine complessivo del sistema e dell’ambiente esterno; l’entità del disordine è espressa dalla funzione di stato entropia S. Sadi Carnot (1824) mostrò che l’efficenza di un motore non può essere del 100%. In ogni processo reversibile, la variazione d’entropia del sistema DS è uguale al rapporto tra calore ricevuto DQ e temperatura assoluta T: DS = DQ/T Quindi in ogni reazione spentanea irreversibile: DS > DQ/T (si applica a tutti i processi reali)
 è un termine coniato da Rudolph Clausius nel")
Entropia l l l Entropia (S) è un termine coniato da Rudolph Clausius nel XIX secolo. L’Entropia la possiamo immaginare come la misura del grado di disordine di un sistema. Clausius era convinto della significatività del rapporto tra il calore ceduto e la temperatura alla quale esso viene ceduto: q T
; l E’")
Ordine vs. Disordine E’ facile convertire energia ordinata in energia termica (disordinata); l E’ difficile convertire energia termica in energia ordinata l Statisticamente, ordine disordine è irreversibile l
 “L’entropia di un sistema isolato")
Seconda legge della termodinamica La legge sul disordine (entropia) “L’entropia di un sistema isolato termicamente non diminuisce mai”
 e l’entalpia (H), l’entropia è una funzione")
Entropia l l Come l’energia totale (E) e l’entalpia (H), l’entropia è una funzione di stato. Quindi, S = Sfinal Sinitial Una funzione di stato è una grandezza fisica o una proprietà di un sistema. Dipende solamente dallo stato iniziale e finale e non dal particolare cammino seguito per arrivarvi. Descrive lo stato di equilibrio di un sistema e quindi, le sue variabili termodinamiche (P, V e T) sono ben definite e non variabili nel tempo.

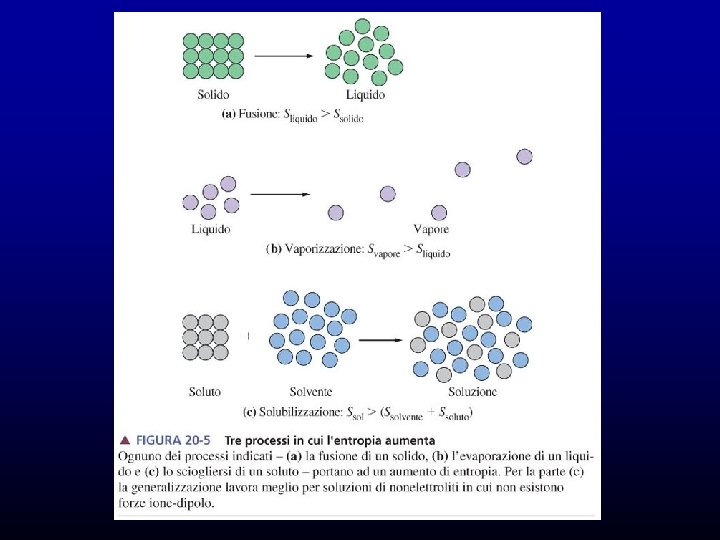
Entropia e II° principio della termodinamica L’entropia S è una grandezza termodinamica che misura il grado di disordine (o della casualità) di un sistema. Tale grandezza è una funzione di stato per cui per un dato processo è possibile definire univocamente una variazione di entropia S = Sf-Si La variazione di entropia per alcuni processi è qualitativamente intuitiva: ad esempio per un processo di fusione si deve avere S>0 poiché il grado di disordine aumenta S>0 Solido cristallino ordinato Liquido disordinato Sf>Si Ad esempio per la fusione di 1 mole di H 2 O si ha S = 22 J/K Le unità dell’entropia sono joule/°K

Un processo avviene naturalmente come risultato di un aumento complessivo del disordine del sistema. Vi è una tendenza naturale dei sistemi a mescolarsi e deteriorarsi o, in generale, a dare un aumento del disordine. Questo concetto è espresso in modo più preciso dal secondo principio della termodinamica Per un processo spontaneo l’entropia totale di un sistema e del suo ambiente (cioè dell’universo) aumenta. Si noti la differenza con il primo principio: l’energia totale rimane costante, mentre l’entropia totale aumenta. È più utile esprimere il secondo principio in modo da riferirsi alle proprietà del sistema considerato più che a quelle di tutto l’universo. A tale scopo prendiamo in esame un sistema in cui ha luogo un dato processo e consideriamo lo scambio di calore tra esso e l’ambiente

Sia q il flusso di calore che entra o esce dal sistema verso l’ambiente: in generale l’entropia accompagna il flusso di calore in quanto tale calore contribuisce a rendere più disordinato il sistema verso cui fluisce. Se il processo avviene in condizioni di equilibrio o quasi equilibrio (reversibile), si può dimostrare che la variazione di entropia del sistema è data da:

Per l’ambiente si ha analogamente Di modo tale che Processo quasi all’equilibrio Se il processo è invece spontaneo si deve avere un aumento dell’entropia nel sistema cioè Processo spontaneo L’ultima equazione può essere considerata come una riformulazione del secondo principio

Variazioni di entropia nelle transizioni di fase Nell’equilibrio tra due fasi lo scambio di calore può avvenire reversibilmente, sono quindi processi quasi all’equilibrio. Nel caso di una transizione di fase alla temperatura Ttrans si ha q = Htrans e il S è dato da trans = Fusione Evaporazione Sublimazione Per la fusione di una mole di acqua a 0°C, con Hfus = 6, 0 k. J, si ha:

Entropia a scala molecolare l l Ludwig Boltzmann descrisse il concentto di entropia a livello molecolare. La temperatura è la misura dell’energia cinetica media delle molecole in un campione sample.

Entropia a scala molecolare l Le molecole hanno vari tipi di movimenti: u u u Translazionale: movimento delle molecole da un ount ad un altro. Vibrazionale: movemnto periodico degli atomi all’interno di una molecola. Rotazionale: rotazione delle molecole attorno ad un’asse o rotazione sui legami .

Entropia a scala molecolare l Boltzmann “bloccò” i movimenti di un campione di molecole ad un particolate tempo t. u l Come scattassimo una istantanea delle molecole. Egli riferì questo campionamento ad un microstato del sistema termodinamico.

Esempi di variazioni di entropia in processi naturali “l’entropia dell’universo tende ad un massimo” – Clausius, 1897
 di carattere")
Termodinamica e geochimica La termodinamica comprende lo studio di grandezze macroscopiche (variabili) di carattere fisico (ad esempio la pressione, ENERGIA) e chimico (ad esempio il potenziale redox). La geochimica studia il comportamento degli elementi chimici (ad esempio l’andamento delle reazioni chimiche).

Termodinamica e geochimica Attraverso le grandezze termodinamiche ed i principi di termodinamica che descrivono le modalità delle variazioni di energia di un sistema, è possibile prevedere l’andamento di reazioni chimiche, il cui motore è lo scambio di energia.
 e possibile definire")
Termodinamica e geochimica Con i parametri termodinamici (temperatura, pressione, ecc. ) e possibile definire lo stato d’equilibrio di un sistema e quindi l’equilibrio di una reazione chimica.

Stati di energia l l l Instabile: caduta o rotolamento Stabile: a riposo nel più basso stato energetico Metastabile: stato energetico intermedio

Applicazioni della termodinamica in geochimica: paragenesi mineralogiche Tramite lo studio degli equilibri chimici di reazioni che avvengono in ambienti profondi (non direttamente osservabili) è possibile ricostruire la chimica dei reagenti (rocce+fuidi) che danno luogo ai prodotti di superficie (fluidi e fasi minerali secondarie).

Applicazioni della termodinamica in geochimica: geotermometria L’espressione dei geotermometri deriva dal calcolo dei rapporti di concentrazione delle specie di una determinata reazione all’equilibrio, effettuato tramite i valori delle costanti termodinamiche ad esse relative

Energia Libera di Gibbs L’energia libera di Gibbs è una misura dell’energia chimica. Dalla combinazione del primo e secondo principio della termodinamica abbiamo: G = H - TS Dove: G = Energia Libera di Gibbs H = Entalpia (contenuto di calore) T = Temperatura in gradi Kelvin S = Entropia

Termodinamica Per una fase possiamo determinare V, T, P, ecc. , ma non G o H Possiamo determinare solo variazioni (cambiamenti) in G o H come variamo altri parametri del sistema Esempio: misurare DH per una reazione per calorimetria - il calore rilasciato o assorbito mentre una reazione procede Assumeremo uno stato di riferimento arbitrario e altrettanto arbitrariamente assegneremo un valore di H ad esso: Selezionato 298. 15 K e 0. 1 MPa (condizioni di lab) …e H = 0 per elementi puri (nel loro stato naturale - gas, liquido, solido) per quello stato di riferimento
 +")
Termodinamica Nel nostro calorimetro, possiamo determinare il DH per la reazione: Si (metallo) + O 2 (gas) = Si. O 2 DH = -910, 648 J/mol = entalpia molare di formazione del quarzo (a 298, 0. 1) Utilizzata come valore standard di H per una fase Entropia ha uno stato di riferimento universale: l’entropia di ogni sostanza = 0 at 0 K, cambia con la temperatura Allora, possiamo usare G = H - TS per determinare la G del quarzo = -856, 288 J/mol

Termodinamica Dal momento che d. H=Vd. P, possiamo anche usare l’equazione, che permette di calcolare d. G anche a condizioni T e P non standard: d. G = Vd. P – Sd. T Possiamo usare quest’equazione per calcolare G per ogni fase a qualsiasi T e P integrando GT 2 P 2 GT 1 P 1 = P 2 SP 1 Vd. P T 2 ST Sd. T 1

Sommario: u u u Termodinamica G è una misura della stabilità chimica relativa per una fase Possiamo determinare G per ogni fase misurando H e S per la reazione che forma la fase in considerazione dai singoli elementi Possiamo determinare G per ogni T e P matematicamente F Più accurato se conosciamo come V e S variano con P e T • d. V/d. P è il coefficiente della compressibilità isotemica • d. S/d. T è la capacità di calore (Cp) Uso? Se conosciamo G per varie fasi, possiamo determinare quale è più stabile F Perchè il fuso è più stabile di un solido a alte T? F A 150 km di profondità è stabile il diamante o la grafite?
 B: fase liquida")
Energia libera e temperatura A: fase solida più stabile (bassa T) B: fase liquida più stabile (alta T) Equilibrio a Teq u GLiq = GSol

Energia libera l l In ogni trasformazione spontanea, a P e T costanti si ha una diminuzione della G del sistema; solo in tal caso si ha un aumento della S complessiva (II° principio); Considerando solo il termine H sarebbero possibili solo i processi esotermici (DH<0). Al contrario, anche i processi endotermici (DH>0) possono essere spontanei, purchè comportino un aumento di S tale che sia TDS>DH

Equilibrio ed energia libera Un sistema sarà all’equilibrio quando la SG di tutti i componenti del sistema sarà la minima possibile. Nel corso di una reazione la variazione di H è lineare, mentre –TS segue un andamento con un minimo (corrispondente al massimo di entropia). Conseguentemente, anche la curva che descrive la variazione di G presenta un minimo, corrispondente alla situazione d’equilibrio.

L’equilibrio chimico All’equilibrio le concentrazioni dei componenti sono costanti, quindi sarà costante il loro rapporto, cosa che si esprime attraverso la legge dell’azione di massa: modificando la concentrazione di un componente, automaticamente si modificano le altre in modo che il rapporto K resti costante. Kc è la cosiddetta costante d’equilibrio
 + O")
L’equilibrio termodinamico In un recipiente a 700°C si pongono 2 SO 2(g) + O 2(g) ⇄ 2 SO 3(g) Quoziente di reazione: Il sistema non è all’equilibrio, ma tende a spostarsi verso Q = K La reazione si sposta verso destra fino a Q = K

L’equilibrio termodinamico Per una qualsiasi reazione: A+ B C + D Si può calcolare il quoziente di reazione, Q, e confrontarlo con K Se Q = K si ha equilibrio Se Q > K la reazione si sposta a sinistra Se Q < K la reazione si sposta a destra fino a Q = K

Equilibri in soluzioni acquose e termodinamica geochimica

Soluzioni acquose Sa lt Na. Cl --> Na+ + Cl- Concentrazione Na+ Saturazione Disequilibrio Modalità attiva Tempo Na. Cl Na+ + Cl. Se disturbiamo in sistema il Principio di Le Châtelier ci dice: Equilibrio Quando una reazione all’equilibrio è disturbata, l’equilibrio si ristabilisce Modalità inattiva contrapponendosi al disturbo.

Ca. CO 3 + 2 HCl --> Ca 2+ + CO 2 + H 2 O + 2 Cl. A contatto con l’atmosfera la CO 2 tende a sfuggire e la reazione non raggiungerà l’equilibrio. La reazione continuerà sino a che uno dei due reagenti finisca. La reazione progredisce a COMPLETEZZA Un altro motivo per cui una reazione non raggiunge l’equilibrio è quando il tasso di una reazione è lento. Queste reazioni sono relative a quelle che coinvolgono la trasformazione di un solido in un altro solido. Cinetica delle reazioni: con velocità di reazione si intende il tasso di variazione nel tempo del grado di avanzamento di una reazione chimica, ovvero il tasso di variazione nel tempo delle concentrazioni delle specie chimiche coinvolte nella reazione.

La Legge di Azione di Massa Claude Louis de Berthollet: la direzione delle reazioni chimiche può essere invertita qualora venga aggiunto un eccesso di uno dei prodotti All’equilibrio: a. A + b. B c. C + d. D Costante di equilibrio K = (C)c(D)d (A)a(B)b Coefficienti molari Quoziente della reazione Non si può derivare dalla CINETICA perchè ogni reazione è il risultato di vari step; mentre è derivabile dalla prima e dalla seconda legge della TERMODINAMICA per reazioni fra gas ideali all’equilibrio.

Il Coefficiente di Attività Per applicare la Legge di Azione di Massa a reazioni fra ioni e molecole in soluzioni acquose si deve sostituire la concentrazione con l’ATTIVITA’: a = c Concentrazione molare Coefficiente di attività I g correggono le concentrazioni molari per le interferenze di altri ioni in soluzioni reali In genere i coefficienti di attività sono inferiori a 1 e quindi: La concentrazione effettiva (attività) degli ioni è inferiore di quella misurata K = [C]c [D]d La differenza fra concentrazione ed a [B]b [A] attività non è importante quando si abbia a che fare con soluzioni diluite. I valori numerici dei coefficienti di attività possono essere calcolati dalla teoria di Debye-Hückel.

Attività l l l Le attività ci permettono di calcolare la concentrazione ‘effettiva’ degli ioni in soluzione Siccome gli ioni, che sono idratati, sono circondati in soluzione da ioni a carica contraria, la loro abilità a reagire con altri ioni (e. g. per formare un precipitato) è ridotta L’Attività è una misura di come venga ridotta la capacità reattiva degli ioni (“concentrazione effettiva”).

Se consideriamo l’immagine dell’ambiente circostante ad uno ione di cloruro ecco come ci apparirebbe: a) una soluzione acquosa molto diluita: Raggio ionico di cloruro idrato Raggio dello ione cloruro Cl- b) una soluzione acquosa più concentrata (~1 M) di Na. Cl Na+ Cl. Na+

a = c Per diluizioni ∞ ; =1 Senza dimensione… ci permette di mantenere così le stesse dimensioni delle concentrazioni Molalità (m), Formalità (F), Molarità (M), Normalità (N) moli/kg(H 2 O) moli/kg(soluzione) moli/L(soluzione) Indipendenti dalla Temperatura eq/L(soluzione) Dipendenti dalla Temperatura 1 g di H 2 O 0 °C = 1. 00013 m. L 100 °C = 1. 04342 m. L E’ necessario conoscere la T °C a cui ci si riferisce 3. 8 °C = 1. 00000 m. L I coefficienti di attività ( ) possono essere calcolati o sono disponibili sottoforma di tabelle standardizzate
![K = [C]c [D]d [A]a [B]b 1) Le attività devono essere espresse in termini](http://slidetodoc.com/presentation_image_h2/52168d4cae75cd2a576ca3251f512a31/image-60.jpg "K = [C]c [D]d [A]a [B]b 1) Le attività devono essere espresse in termini")
K = [C]c [D]d [A]a [B]b 1) Le attività devono essere espresse in termini di moli ma devono essere riferite ad un volume, kg di soluzione, ecc. 2) Le attività di solidi e dell’acqua sono pari a 1; 3) Le concentrazioni dei gas sono espresse come pressioni parziali in bar; 4) Le reazioni, se non indicato diversamente, sono riferite a 25 °C e 1 bar moli: si usa in quanto le reazioni chimiche risultano dalle interazioni di ioni individuali o molecole il cui numero per mole è definito dal Numero di Avogadro solidi e acqua: il fatto che la loro attività sia 1 per definizione fa si che si possano non considerare nella Legge di Azione di Massa Tuttavia per soluzioni molto concentrate l’attività dell’acqua può essere < 1 Le costanti di equilibrio variano con la temperatura e quindi, questa deve essere specificata. Gli effetti della pressione possono essere trascurabili per condizioni vicine alla superficie

Prodotto di Solubilità La solubilità di un minerale è governata dal PRODOTTO DI SOLUBILITA’, la costante di equilibrio per una reazione del tipo: Ca. SO 4(anidrite) Ca 2+ + SO 42 Il Prodotto di Solubilità è dato da: Se l’anidrite è un solido puro, allora a. Ca. SO = 1. 4 e in soluzioni diluite: a. Ca 2+ (Ca 2+) e a. SO 42 - (SO 42 -).
![Indice di Saturazione In una soluzione naturale, non è detto che [Ca 2+] =](http://slidetodoc.com/presentation_image_h2/52168d4cae75cd2a576ca3251f512a31/image-62.jpg "Indice di Saturazione In una soluzione naturale, non è detto che [Ca 2+] =")
Indice di Saturazione In una soluzione naturale, non è detto che [Ca 2+] = [SO 42 -], poichè ci può essere più di una sorgente per ognuno di questi ioni. In questo caso, si usano gli INDICI DI SATURAZIONE per determinare se un’acqua è satura rispetto, ad esempio, all’anidrite: KSP = 10 -4. 5 [Ca 2+]eq[SO 42 -]eq IAP = [Ca 2+]act[SO 42 -]act Prodotto Ionico di Attività Indice di Saturazione [ ] = Ca 2+ 2 SO act 4 K SP act IAP = K SP

Supponiamo che un’acqua contenga 5 x 10 -2 mol/L Ca 2+ e 7 x 10 -3 mol/L SO 42 -. E’ quest’acqua satura rispetto all’anidrite (Ca. SO 4(s))? Ca. SO 4 Ca 2+ + SO 42 KSP = 10 -4. 5 IAP = (5 x 10 -2)(7 x 10 -3) = 3. 5 x 10 -4 = 10 -3. 45/10 -4. 5 = 101. 05 = 11. 22 > 1, i. e. , IAP > KSP, la soluzione è sovrassatura e l’anidrite dovrebbe precipitare Se = 1, i. e. , IAP = KSP, la soluzione sarebbe satura (condizioni di equilibrio) Se < 1, i. e. , IAP < KSP, la soluzione sarebbe sottosatura; il minerale si dovrebbe solubilizzare.

L’effetto dello ione a comune Le acque naturali sono sistemi complessi e possono essere saturate simultaneamente in diverse fasi mineralogiche. Esempio: quali sono le concentrazioni delle specie in una soluzione in equilibrio sia con barite che con gesso? 1) L’espressione della Legge di Azione di Massa ci dice: Ca. SO 4· 2 H 2 O Ca 2+ + SO 42 - + 2 H 2 O, KSP = [Ca 2+][SO 42 -] = 10 -4. 6 Ba. SO 4 Ba 2+ + SO 42 -, KSP = [Ba 2+][SO 42 -] = 10 -10. 0
![[Ca 2+][SO 42 -] = 10 -4. 6 [Ba 2+][SO 42 -] = 10](http://slidetodoc.com/presentation_image_h2/52168d4cae75cd2a576ca3251f512a31/image-65.jpg "[Ca 2+][SO 42 -] = 10 -4. 6 [Ba 2+][SO 42 -] = 10")
[Ca 2+][SO 42 -] = 10 -4. 6 [Ba 2+][SO 42 -] = 10 -10. 0 Eliminiamo [SO 42 -] sostituendolo con 10 -4. 6/[Ca+]: [Ba 2+] • 10 -4. 6/[Ca 2+] = 10 -10. 0 Bilancio di massa: [Ba 2+] + [Ca 2+] = [SO 42 -]

Le soluzioni acquose che analizziamo sono il risultato di: - processi di dissoluzione congruente e incongruente; - processi di solubilizzazione e saturazione; - processi di natura cinetica; - processi di natura batterica; - processi di solubilizzazione di fasi gassose; - Effetti di temperatura e pressione - etc. TERMODINAMICA E MODELLIZZAZIONE GEOCHIMICA

Come varia K al variare della temperatura? esotermica Nel corso di una reazione esotermica un aumento della T provoca una diminuzione della K, ovvero se con il procedere della reazione si sviluppa calore, un aumento della T dall’esterno causerà una regressione della reazione. endotermica Nel corso di una reazione endotermica un aumento della T provoca un aumento della K, ovvero se con il procedere della reazione viene assorbito calore, aumento della T dall’esterno causerà un avanzamento della reazione.

Come varia K al variare della temperatura? La legge dei gas perfetti dice che: DG 0 = RTlg. K Legge di van’t Hoff termine dipendente dalla temperatura termine non dipendente dalla temperatura

Geotermometria Il legame tra costante d’equilibrio di una reazione e temperatura offre l’opportunità di costruire i geotermometri.

Limiti dei calcoli geotermometrici: I dati termodinamici sorgente dei dati deve essere omogenea e possibilmente recente variazioni con T e P Øampie variazioni di T per piccole variazioni composizionali Øminima influenza della pressione

Limiti dei calcoli geotermometrici Cinetica d’equilibrio “Ogni reazione chimica è caratterizzata da una differente cinetica” Il completo equilibrio di reazione si stabilisce in un tempo indefinito Reazioni di tipo differente riflettono equilibri relativi a diverse condizioni T e P di uno stesso sistema Le temperature calcolate dipendono “quasi” sempre dal geotermometro adottato

Termodinamica e cinetica l l La termodinamica esamina le condizioni che devono essere soddisfatte perché una certa reazione avvenga spontaneamente; La cinetica chimica esamina i fattori che influiscono sul tempo necessario perché una reazione giunga a completezza.

Fattori che influenzano la cinetica l l l Natura dei reagenti; Concentrazione dei reagenti; Temperatura; Catalizzatori; Area di contatto tra le fasi.

Espressione della velocità di reazione V = d. C/dt V = velocità della reazione V assumerà valori negativi per un reagente, positivi per un prodotto C = concentrazione della specie t = tempo Al procedere della reazione DC diminuisce progressivamente, tendendo a zero.

Velocità ed energia La ragione della diversa velocità delle reazioni risiede nell’energia coinvolta nel processo. Per passare dall’energia dei reagenti (energia iniziale del sistema) a quella dei prodotti (energia finale del sistema) occorre superare una barriera di potenziale, necessaria per la formazione di un complesso attivato (intermedio di reazione) caratterizzato dall’energia di attivazione. Si descrive, cioè, un profilo energetico di reazione. La velocità della reazione è proporzionale al numero di molecole che hanno l’energia sufficiente per superare la barriera di potenziale. La distribuzione dell’energia tra molecole dello stesso tipo è espressa dalla legge di Maxwell-Boltzmann.

Legge di Maxwell-Boltzmann E’ una legge sperimentale che rappresenta il numero di particelle che possiedono una certa energia, in funzione dell’energia stessa; cioè, ad ogni valore di energia (ad una data T) corrisponde un numero definito di particelle con quella energia. All’aumentare di T aumenta il numero di molecole con E sufficiente a superare la barriera energetica, perciò aumenta la velocità di reazione L’azione dei catalizzatori consiste nel ridurre la barriera di potenziale, in pratica l’energia di attivazione del processo

Termodinamica statistica La relazione tra la temperatura assoluta T del gas e l’energia cinetica media delle particelle si esprime come: , dove KB=R/NA=1, 38 10 23 J/°K è la cosiddetta costante di Boltzmann, con NA = 6, 02 1023 il numero di Avogadro, cioè il numero di molecole in una mole. Si introduce il concetto di legge di probabilità La meccanica statistica (o termodinamica statistica), a differenza della Termodinamica classica, studia le proprietà dei sistemi da un punto di vista microscopico e si propone di derivare e generalizzare le leggi termodinamiche.

Termodinamica statistica Condizioni perché avvenga una reazione: le molecole devono “incontrarsi”, o “urtarsi” l’urto deve essere “efficace” le molecole devono avere energia abbastanza alta 1) Frequenza di urto in fase gassosa circa 1028 s-1 cm-3 (e dipendenza quadratica dalla temperatura) Sezione d’urto: He 0. 13 H 2 0. 15 CO 2 0. 66 Ne 0. 17 N 2 0. 31 H 2 O 0. 23 Ar 0. 26 O 2 0. 27 Hg 0. 41 Kr 0. 32 Cl 2 0. 43

Termodinamica statistica Sezione d’urto: quali sono le reali dimensioni di un atomo, o di una molecola? Particella gassosa con velocità v, tutte le altre ferme. . area d’urto d vt nel tempo t la particella in movimento “spazza” un volume se la densità è il numero di urti è tenendo conto che anche le altre si muovono, nell’unità di tempo il numero di urti è
 è Non tutti")
Termodinamica statistica La velocità media (e quindi la frequenza degli urti) è Non tutti gli urti sono efficaci Br B r f: frazione degli urti che avvengono con l’orientazione adatta Br Br non efficace Nel caso di soluzioni liquide (o di solidi), si parla di diffusione gli “incontri” sono molto più rari, ma durano di più (effetto gabbia del solvente) Quale è la velocità di diffusione di una specie in soluzione?

. . . e il coefficiente di diffusione è In soluzione l’orientazione in genere non è un problema perché le molecole rimangono vicine più a lungo. Sia in gas che in soluzione, c’è un fattore energetico solo le molecole con E > Ea possono reagire velocità Origine della barriera energetica: superfici di energia potenziale, coordinata di reazione. . Quindi, velocità di reazione A+B P
 Portando in diagramma il")
Equazione di Arrhenius lg. Kv = lg. A-Ea/2. 303 R(1/T) Portando in diagramma il lg decimale della KV di velocità contro 1/T, determinando perciò le K a diverse temperature, è possibile determinare l’energia d’attivazione Ea di una reazione mediante il calcolo del coefficiente angolare (pendenza della retta). Ea = energia di attivazione A = cost. di reazione KV = cost. di velocità T = temp. assoluta
- Slides: 82