Tecniche di trattamento del DNA Libreria genomica Estraendo

Tecniche di trattamento del DNA

Libreria genomica Estraendo il materiale genetico dalle cellule di un individuo e trattandolo con uno o più enzimi di restrizione, si ottengono numerosi frammenti di diverse misure. Miscelati con i plasmidi vettori (sottoposti allo stesso trattamento enzimatico) è possibile ottenere una serie di DNA ricombinanti, ognuno dei quali può essere integrato in batteri differenti. Ogni batterio integrerà un determinato plasmidio vettore e, replicandosi, darà origine ad una colonia fatta da batteri-cloni contenenti un determinato frammento di DNA. In yal modo i frammenti vengono amplificati. L’insieme delle colonie formerà una libreria genomica La libreria si dirà rappresentativa se, effettivamente, contiene tutti i frammenti del materiale genetico originario e, nell’insieme, costituiscono l’intero genoma dell’individuo, quindi nessuna informazione è stata persa (non sempre, infatti, i risultati di questa tecnica sono ottimali). Su tale libreria, attraverso tecniche di «blotting» , blotting si potrà individuare e isolare il singolo gene d’interesse.
 complementare Il materiale genetico esogeno da utilizzare nelle biotecnologie, solitamente,")
c. DNA (DNA complementare) complementare Il materiale genetico esogeno da utilizzare nelle biotecnologie, solitamente, non è estratto direttamente da molecole di DNA. Pur contenendo il gene target, infatti, quest’ultimi hanno anche sequenze introniche, introniche non utilizzabili per la traduzione in polipeptidi specifici d’interesse. Per tal motivo si ricorre alla tecnica del c. DNA: 1. Estrazione dell’m. RNA Ø Si frammentano le cellule per liberare gli acidi nucleici Ø L’estratto ottenuto si versa su una resina di agarosio su cui sono agganciati frammenti di DNA sintetico fatto da sequenze singole di deossiribotimidina (Poly-T) Ø Al lavaggio, tutto il materiale viene asportato via ad eccezione delle molecole di m. RNA che rimangano attaccate ai frammenti di DNA sintetico grazie alle code Poly-A. Ø Con aggiunta di acqua, o apposite soluzioni, le molecole di m. RNA si staccano dal polimero di agarosio.
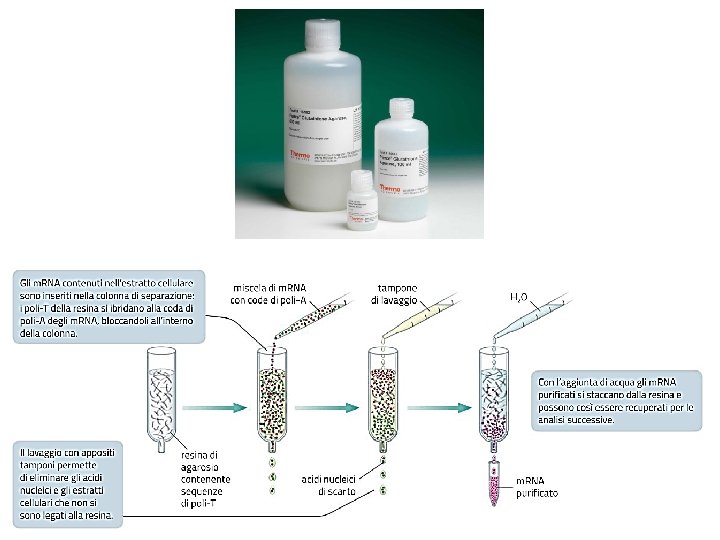

2. Sintesi di c. DNA Ø All’m. RNA purificato si aggiungono nuovamente frammenti poly-T che, legandosi alle code Poly-A, questa volta funzionano da primers per l’enzima trascrittasi inversa Ø Si aggiungono la trascrittasi inversa e i nucleotidi liberi Si costruirà in tal modo la prima catena del c. DNA. Ø Si rimuove dalla miscela l’m. RNA. La Trascrittasi inversa continuerà il suo lavoro sintetizzando la seconda catena di c. DNA. Alla fine si otterrà una miscela di c. DNA corrispondenti alla miscela di m. RNA iniziale. m. RNA - DNA filamento singolo c. DNA

Librerie di c. DNA Il materiale, estratto e purificato con la tecnica precedente, contiene tutte le molecole di c. DNA dei geni attivi, cioè espressi dalla cellula. Con essi, attraverso DNA ricombinante, si potrà costruire una libreria di c. DNA La differenza tra la libreria genomica e quella c. DNA è che la prima contiene tutto il corredo genetico, comprensivo degli introni, mentre la seconda contiene le informazioni «sgrezzate» di tutti i geni espressi dalla cellula nel momento in cui si è «trattata» .

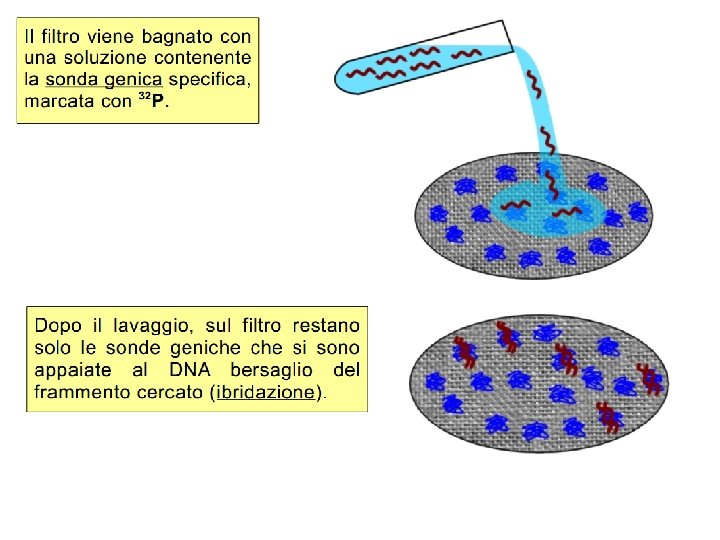
Individuazione gene interessato: le sonde geniche In una libreria di c. DNA o genomica vi si trovano miglia di geni differenti, indistinguibili l’uno dall’altro. Per individuare un gene specifico si utilizza la tecnica della «ibridazione» attraverso una sonda genica Una sonda genica è una sequenza corta di nucleotidi (alcune decine), sintetizzata in laboratorio e corrispondente ad un tratto del gene, da individuare nella libreria, di cui si conosce bene la sequenza nucleotidica. Tale sequenza la si può conoscere dall’analisi chimica della struttura primaria del polipeptide codificato dal gene, oppure tramite confronto con sequenze già note di un gene analogo, ma di altre specie. Le sonde geniche sono «marcate» marcate con isotopi radioattivi o con molecole fluorescenti ovvero colorate.

Un metodo di utilizzo delle sonde geniche è quello del Colony Blot

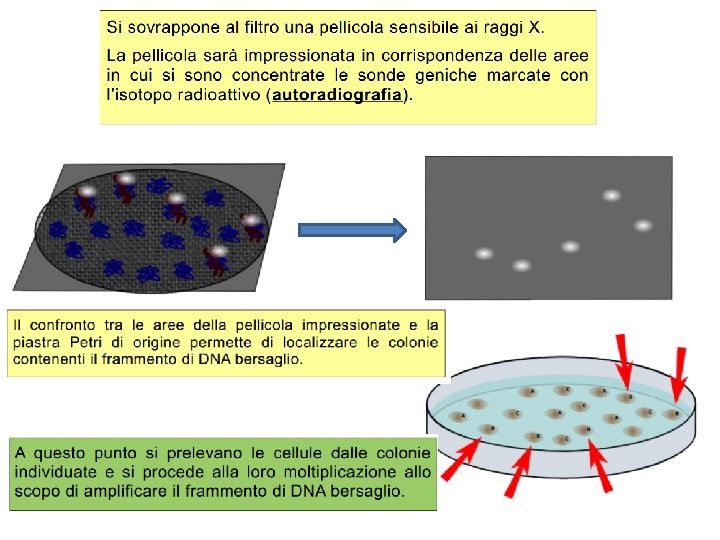
Semina dei batteri. I batteri di una libreria si seminano su un terreno di coltura solido (agar) Piastra di Petri Il filtro «adsorbirà» una parte di cellule per ogni colonia (a «stampo) Denaturazione del DNA: si scalda il disco a 95°. A tale temperatura si rompono i legami idrogeno tra le basi complementari e le due catene si separano.

Southern Blotting E’ possibile individuare ed isolare un frammento di DNA, contenente un gene specifico partendo direttamente da materiale genetico estratto da un tessuto (se se ne un buon quantitativo, solitamente dopo tecniche di amplificazione). Southern Blotting Dopo frammentazione con enzima di restrizione, il materiale genetico si sottopone a elettroforesi su gel di agarosio (polisaccaride naturale) o di poliacrilammide (polimero di sintesi). I vari frammenti, sottoposti a campo elettrico (elettroforesi), elettroforesi migrano verso il polo positivo (Il DNA, essendo acido per i gruppi fosfato, è negativo). La velocità sarà diversa per i vari frammenti ( i più piccoli saranno più veloci). Questo porterà alla separazione dei frammenti stessi.

Il gel viene immerso in una soluzione alcalina per denaturare il DNA (separare la doppia catena); solitamente si tratta di una soluzione molto diluita di Na. OH per un tempo pari a 15 minuti. Dopo denaturazione, il gel viene coperto da un foglio di nitrocellulosa o nylon a carica positiva e, sopra di questo, viene posta una pila di fogli assorbenti I frammenti di DNA verranno adsorbiti, a stampo, su questi fogli.

Il foglio di nitrocellulosa viene immerso in una soluzione contenente una sonda genica marcata in vario modo (fluorescenza, radioattività, ecc. . ) che ibridizza con le sequenze di DNA complementari presenti sul foglio, permettendone l’individuazione. L’individuazione viene fatta dopo lavaggio della nitrocellulosa, grazie ad una lastra fotografica che metterà in evidenza le sonde legate ai frammenti target. Una variante a questa tecnica è il Northern Blot: Blot viene fatta su RNA

PCR

Polymerase Chain Reaction La tecnica del colony blot e del clonaggio batterico permettono di identificare e clonare un gene (o, in genere, un determinato frammento di DNA), tuttavia non si dispone di quest’ultimo in maniera isolata, pura. Nel 1983, è stata ideata una tecnica biochimica in grado di individuare, individuare isolare e amplificare un gene specific: la PCR (Polymerase Chain Reaction). Ingredienti base: – DNA o c. DNA – 2 primers specifici, complementari tra loro, sintetizzati in laboratorio prendendo come stampo alcune sequenze di nucleotidi situati a monte del gene “target”. – DNA polimerasi termostabile (Taq polimerasi) – I 4 tipi di desossiribonucleotidi

I cicli della PCR La tecnica prevede 3 fasi da ripetere in 30– 35 cicli: 1. Denaturazione (95°C), 10 -40 sec separazione catene 2. Annealing (ibridazione) (50– 65°C), 30 -120 sec legame dei primers ai siti specifici. 3. Polimerizzazione (68 -72°C), il tempo dipende dalla lunghezza del frammento sintesi nuove catene Dopo 30 cicli si avranno: • 2 x 30 catene prolungate (molecole contenenti il gene specifico + diversi altri geni) • 230 catene «target» (molecole del gene specifico)

Lo strumento utilizzato per la PCR si chiama «termociclatore» . termociclatore

Sequenziamento di Sanger A metà circa degli anni ’ 70 Frederick Sanger introdusse la prima tecnica per il sequenziamento del DNA, ovvero per la determinazione della successione di nucleotidi in frammenti di DNA.

Tecnica di Sanger 1 Si parte da molecole di DNA replicate tramite tecnica PCR o tramite clonaggio in plasmidi di E. Coli 2 Ottenuta una certa quantità di molecole di DNA, si purificano e si trattano al calore (denaturazione a 95° in modo da ottenere catene singole

3 Il materiale ottenuto si distribuisce equamente in 4 provette 4 In ognuna si aggiunge una pari quantità dei 4 nucleotidi liberi
,")
5 In ciascuna provetta si aggiunge uno specifico tipo di di-desossiribonucleotide (dd. Nt. P), dd. Nt. P cioè un desossiribonuclotide mancante di un ossigeno in posizione 3’ (oltre che in posizione 2’) dd. ATP dd. GTP dd. TTP d. Nt. P dd. CTP 6 In ciascuna provetta si aggiunge il complesso enzimatico DNA-polimerasi corredato dal suo primer (sequenza di nucleotidi che agganciandosi in modo complementare all’inizio della catena di DNA permette l’attacco dell’enzima)

7 La DNA polimerasi inizierà la «costruzione» di nuove catene di DNA, a stampo su quelle originali presenti, utilizzando i nucleotidi liberi a disposizione.

La DNA polimerasi non distinguerà i d. NTP dai dd. NTP, per cui utilizzerà indifferentemente e casualmente l’uno o l’altro. Il «lavoro» di polimerizzazione proseguirà finchè, casualmente, l’enzima non avrà incontrato e incorporato nella catena un dd. NTP. A tal punto la catena si interromperà in quanto non sarà stato possibile formare il legame fosfoesterico in posizione 3’ (mancando il gruppo -OH per la «condensazione» ). Dato il fatto che un determinato nucleotide si trova ripetuto svariate volte lungo la catena di acido nucleico, data, inoltre, la casualità dell’interruzione, di ogni singola molecola di DNA si duplicherà un solo frammento di lunghezza casuale Dato l’altissimo numero di molecole in una provetta ci sarà la miscela dei frammenti duplicati di tutte le lunghezze possibili. Tali frammenti termineranno, comunque, con lo stesso dd. NTP (per esempio, come in figura, il dd. C). dd. C

Dato che ogni provetta contiene un solo tipo di dd. NTP, dd. NTP i frammenti in ognuna di esse saranno caratterizzati da nucleotidi terminali differenti. Con le miscele così ottenute, si allestisce un’analisi elettroforetica presentante 4 pozzetti in ognuno dei quali si appoggia una goccia di ciascuna provetta. Alla fine dell’esame (trascinamento elettroforetico), elettroforetico si avrà un grafico con bande in diverse posizioni (a seconda della grandezza dei frammenti in gioco) da cui si ricava la sequenza di nucleotidi nella molecola di DNA originaria.


 (es. quella di")
Oggi esistono tecniche più sofisticate definite di Next Generation Sequencing (NGS) (es. quella di high-throughput sequencing) sequencing che consentono di analizzare un'elevatissima quantità di sequenze di DNA di un soggetto (anche di soggetti diversi, in parallelo) in breve tempo e a costi relativamente contenuti. Se l'output giornaliero di un sequenziatore con tecnica Sanger è nell'ordine delle migliaia di basi, l'output delle macchine di high-throughput sequencing è nell'ordine delle gigabasi.
- Slides: 29