Taller 7 Sndromes Hereditarios con Afectacin Cutnea Dra









 : lesiones maculares,")







")











 67")
 67")
 67")












")










- Slides: 64
Taller 7: Síndromes Hereditarios con Afectación Cutánea Dra. Begoña Graña Suárez Servicio de Oncoloxía Médica Unidade de Alto Risco en Cáncer e Asesoramento Xenético Hospital Arquitecto Marcide Area Sanitaria de Ferrol- A Coruña begona. grana. suarez@sergas. es
DEFINICIONES Genodermatosis: enfermedades cutáneas de origen genético. Facomatosis Grupo de enfermedades de origen hereditario que se caracteriza por provocar deformaciones congénitas derivadas del ectodermo en diversas áreas del cuerpo, sobre todo del sistema nervioso central. Síndromes hereditarios oncológicos y dermatológicos Enfermedades hereditarias con manifestaciones cutáneas que se asocian a la predisposición a padecer cáncer cutáneo y/o en otros órganos
Una familia es SOSPECHOSA y puede precisar consejo genético si: - Aparecen casos a edad tempranas inusuales, - Cáncer multifocal en un órgano o bilateral (si organo par), - “Cluster individual”, 2 o + tumores sincrónicos o metacrónicos, - “Cluster familiar” compatible con un síndrome reconocible o no, - Evidencia de transmisión autosómica dominante (penetrancia), - Cáncer en individuos con anomalías congénitas , signos cutáneos…
NEUROFIBROMATOSIS I Esclerosis tuberosa Xeroderma pigmentosum Melanoma Familiar Sindrome de Gorlin Síndrome de Gardner Síndrome de Birth Hogg Dubbé Síndrome de Muir Torre Síndrome de Cowden Progeria
XERODERMA PIGMENTOSSUM
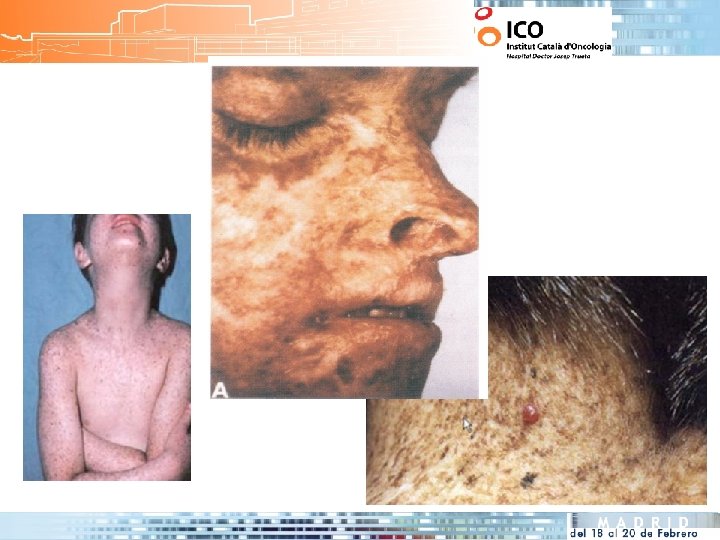
• Herencia Autosómica Recesiva • Incidencia 1: 1. 000 • Penetrancia 100% • Heterogenicidad genética: 8 genes diferentes conocidos (XPA->XPG+variante) Manifestaciones clínicas 1. Cutáneas: fotosensibilidad extrema. Envejecimiento prematuro de la piel fotoexpuesta (labios, ojos, boca y lengua). Eritema, alteracion pigmentación, atrofia, telangiectasias, Alta incidencia de cáncer en dichas áreas 2. Oftalmológicas: Lesiones por exposición UV en conjuntiva y cornea: opacificación, keratitis, conjuntivitis, cáncer conjuntiva… 3. Neurológicas: retraso psicomotor. Microcefalia. Neuropatía axonal o mixta. Sordera progesiva. Deben identificarse lesiones en los 3 sistemas
57 PULMO 59 62 87 <40 66 LH 50 S. CUSHING HIPERPLASIA SUPRARENAL 4 34 32 23 -31 70 2 37 XP 34 ESCAMÓS CUTANI 16 BASOCEL·LULAR 39 XP AMAUROSIS D >70 3 Gi-XPS 1 16/01/04 Casadevall JR, Graña-Suárez B, Hernandez-Yagüe X, Ribera JV, Grasa OH, Vidal JB Eur J Dermatol. 2008 Dec 23.
MELANOMA FAMILIAR
Melanoma sobre nevus displásico • Diagnóstico clínico: NEVUS ATÍPICOS (o DISPLÁSICOS) : lesiones maculares, pigmentación variable, bordes irregulares con un diámetro ≥ 5 mm. Pueden aparecer en zonas no fotoexpuestas. MELANOMA MALIGNO: son signos de sospecha de transformación a melanoma el aumento del diámetro, cambios en los bordes y en la pigmentación, así como prurito.
Diagnóstico • Características diferenciales del melanoma maligno familiar y esporádico: FAMILIAR ESPORÁDICO Antecedentes familiares ≥ 2 primer grado NO Edad media al diagnóstico 35 años [29 -36] 54 años [50 -57] Diagnóstico <20 años 10% 2% Nevus displásicos En la mayoría de los casos 30% Nº de melanomas primarios (>30%) 4%
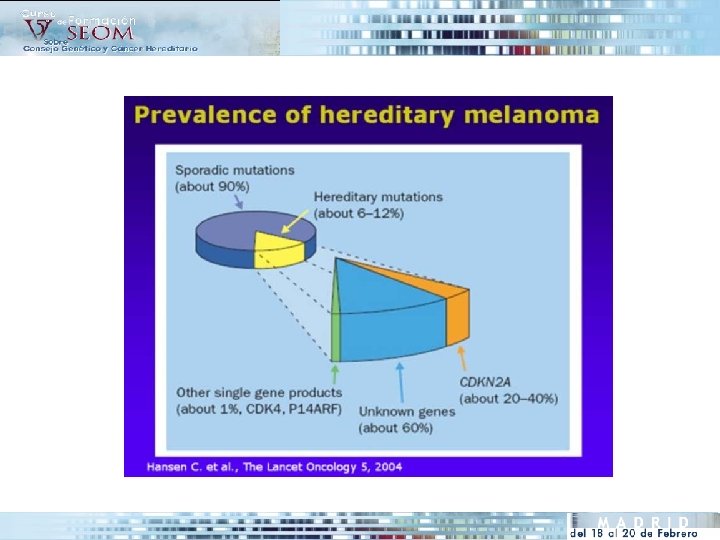
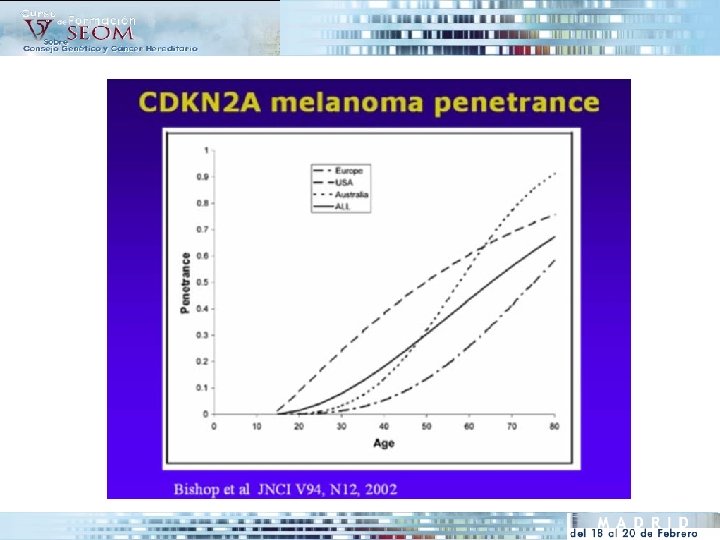
• El melanoma familiar es genéticamente heterogéneo, sigue un patrón de Genes involucrados herencia autosómica dominante con una penetrancia incompleta. • Existen varios genes involucrados en su desarrollo: GEN MECANISMO CROMOSOMA COMENTARIOS ? ? 1 p 36 Asociación con nevus malignos CDKN 2 A Supresor tumoral 9 p 21 Existe una asociación con cáncer de páncreas CDK 4 Oncogén 12 q 14 Se han reportado solo 2 familias Otros 6 p 6 q 10 q • CDKN 2 A: Desempeña un papel prioritario en el control del ciclo celular La probabilidad de encontrar mutación (25%) depende del número de afectos en la familia – 5% ( si 2 miembros afectos de melanoma) – 20 -40% (≥ 3 miembros) – 100% (≥ 13 miembros)
Manejo de Individuos de alto riesgo -Autoexploración cutánea 1 vez al mes -Examen clínico cada 6 meses -Iniciar exámen cutáneo antes de los 10 años -Toma de fotografías basales de los nevus atípicos -Intensificar vigilancia si fluctuación hormonal -Biopsia excisional de las lesiones sospechosas -Uso de reglas métricas junto a las lesiones -Evitar la exposición solar El papel de las pruebas genéticas no está bien definido en familias de riesgo. Según el Melanoma Genetics Consortium es prematuro ofrecer estas pruebas fuera de protocolos de investigación definidos y tan sólo después de un adecuado consejo genético, dada: -Baja probabilidad de encontrar mutaciones (25%) -Incertezas sobre la penetrancia (incompleta y variable) -Falta probada en estrategias de prevención y vigilancia Las medidas de prevención van encaminadas a evitar factores de riesgo y a un diagnóstico precoz
61 >80 >60 88 3 76 68 61 62 61 MELANOMA 59 PROS 36 5 32 2 3 48 CEREBRAL 30 EXTIRPACIÓN: 3 lunares (NO AP) 1 2 Gi-CDK 1 27/10/05
61 >80 >60 88 3 76 68 61 62 61 MELANOMA 59 PROS CDKN 2 A G 101 W 36 5 32 2 3 48 CEREBRAL 30 EXTIRPACIÓN: 3 lunares (NO AP) 1 2 Gi-CDK 1 27/10/05
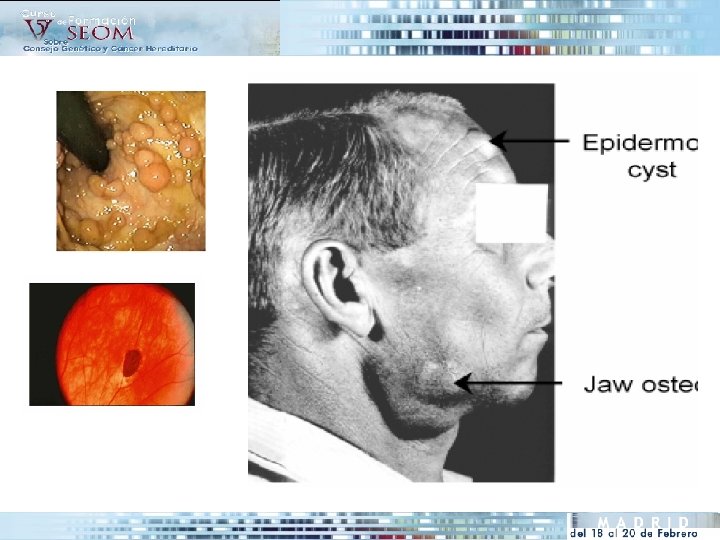
SÍNDROME DE GORLIN (síndrome del carcinoma basocelular nevoide)
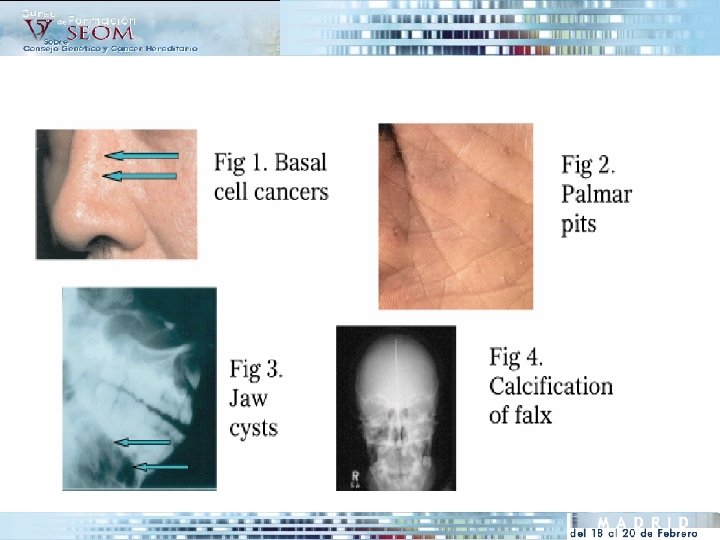
• Herencia Autosómica Dominante • Incidencia 1: 40. 000 • Penetrancia 100% • Etiología: mutaciones germinales en el gen PTCH (9 q 22. 3) identificables hasta en el 60 -85% de los pacientes. 30% “de novo” Criterios Mayores Criterios Menores 1. >2 carcinomas basocelulares o 1 en < 20 años 1. Macrocefalia 2. Queratoquistes maxilares demostrados en AP 2. Malformaciones faciales (fisura palatina, hipertelorismo, etc …. 3. Tres o mas fositas (“pits”) palmares o plantares 4. Calcificación bilaminar de la hoz del cerebro 3. Otras. Deformaciones esqueléticas 5. Costillas bífidas, fusiondas o expandidas 4. Fibroma de ovario 6. Familiar en primer grado con Síndrome de Gorlin 5. Meduloblastoma (5% de afectos de Gorlin – 12% meduloblastomas en afectos Gorlin Debe cumplirse 2 criterios mayores o 1 mayor y dos menores
19 LH 72 72 BASOCEL 49 47 17 ESCLEROSIS MULTIPLE 16 76 44 46 26 BASOCEL·LURAR 6 -7 QUISTES SEBACIS 51 QUISTES SEBACIS? 21 NO HI HA RELACIO BASOCEL 1 10/11/06
SÍNDROME DE GARDNER
• Herencia Autosómica Dominante • Penetrancia 100% • Etiología: mutaciones en el gen APC (5 q) • 30% de los casos son “ de novo” CRITERIOS DIAGNÓSTICOS PAF 1. Más de 100 pólipos adenomatosos colorrectales SÍNDROME DE GARDNER (variante de la PAF clásica) PAF + lesiones extraintestinales: • Tumores desmoides • Osteomas • Dientes supernumerarios • Hipertrofia Congénita del epitelio Pigmentaio de la retina (CHRPE) • Quistes epidérmicos
95 >70 <50 70 72 66 75 34 Colono 2004 3 50 CCR 72 35 Colono 2004 10 62 44 CCR 45 43 CCR 33 Colono 2004 10 64 68 60 MAM >70 32 CCR 8 8 Colono - Gi-POL 4 27/05/05
95 >70 <50 70 72 66 75 34 Colono 2004 3 50 CCR 72 45 43 CCR 35 Colono 2004 10 33 S. GARDNER QUIST EPIDERMOIDES MALFORMACIONS DENTALS PÒLIPS GÀSTRICS 32 CCR 8 62 44 CCR 64 68 60 MAM >70 POLIPOSI 14 PÒLIPS 8 Colono - Gi-POL 4 27/05/05
95 >70 <50 70 72 66 75 34 Colono 2004 3 50 CCR 72 45 43 CCR 35 Colono 2004 10 33 S. GARDNER QUIST EPIDERMOIDES MALFORMACIONS DENTALS PÒLIPS GÀSTRICS 32 CCR 8 Gi-99 APC 1263 del. GGins. AA 62 44 CCR 64 68 60 MAM >70 POLIPOSI 14 PÒLIPS 8 Colono - Gi-POL 4 27/05/05
95 >70 <50 70 72 66 75 34 Colono 2004 Gi-175 3 50 CCR 72 45 43 CCR 35 Colono 2004 Gi-172 10 33 Colono 2004 Gi-176 10 33 S. GARDNER QUIST EPIDERMOIDES MALFORMACIONS DENTALS PÒLIPS GÀSTRICS 32 CCR 8 Gi-99 APC 1263 del. GGins. AA 62 44 CCR 64 68 60 MAM >70 POLIPOSI 14 PÒLIPS 8 Colono - Gi-POL 4 27/05/05
95 >70 <50 70 72 66 75 34 Gi-175 APC WT 3 50 CCR 72 35 Gi-172 APC WT 10 45 43 CCR 33 Gi-176 APC WT 10 33 S. GARDNER QUIST EPIDERMOIDES MALFORMACIONS DENTALS PÒLIPS GÀSTRICS 32 CCR 8 Gi-99 APC 1263 del. GGins. AA 62 44 CCR 64 68 60 MAM >70 POLIPOSI 14 PÒLIPS 8 Colono - Gi-POL 4 27/05/05
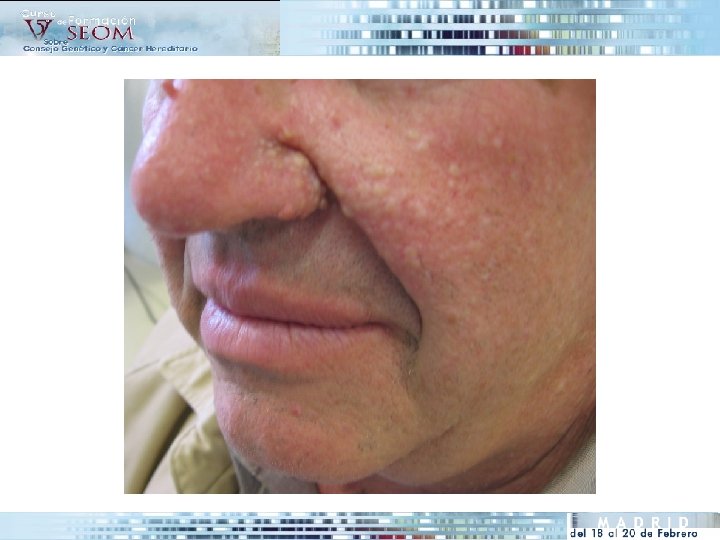
SÍNDROME DE BIRT-HOGG-DUBÉ
• Herencia Autosómica Dominante • Incidencia ? ? ? • Etiología: mutaciones en el gen de la foliculina FLCN (17 p 11. 2). CRITERIO DIAGNÓSTICO 1. Más de 10 lesiones cutáneas compatibles con fibrofoliculomas, tricodiscomas y acrocordones (al menos un fibrofoliculoma histológicamente confirmado) CRITERIOS DE SOSPECHA (2+3): 2. Tumores renales múltiples y bilaterales, principalmente oncocitomas o carcinomas cromófobos 3. Neumotórax espontáneo familiar Criterio 1 de confirmación. Criterios 2 y 3 de sospecha
>70 >60 >70 BHD >60 <40 OVA >65 >55 >70 CCR (NO BHD) 67 BHD ? 70 BHD ORL >70 BHD ? >65 BHD 5 35 BHD 17 PNEUMOTÒRAX 9 43 BHD FIBROFOLICULOMES 19 BHD 18 46 BHD 30 PNEUMOTÒRAX 18 52 BHD OVA BHD Gi-BHD 1 28/10/05
>70 >60 >70 BHD >60 <40 OVA >65 >55 >70 CCR (NO BHD) 67 BHD ? 70 BHD ORL >70 BHD ? >65 BHD 5 35 BHD 17 PNEUMOTÒRAX Gi-128 43 BHD FIBROFOLICULOMES 46 BHD 30 PNEUMOTÒRAX 52 BHD 1733 del. C 9 19 BHD 18 18 OVA BHD Gi-BHD 1 28/10/05
>70 >60 >70 BHD >60 <40 OVA >65 >55 >70 CCR (NO BHD) 67 BHD ? 70 BHD ORL >70 BHD ? >65 BHD 5 35 BHD 17 PNEUMOTÒRAX Gi-128 43 BHD FIBROFOLICULOMES BHD 1733 del. C 46 BHD 30 PNEUMOTÒRAX 52 BHD 1733 del. C 9 19 BHD 1733 del. C 18 18 OVA BHD Gi-BHD 1 28/10/05
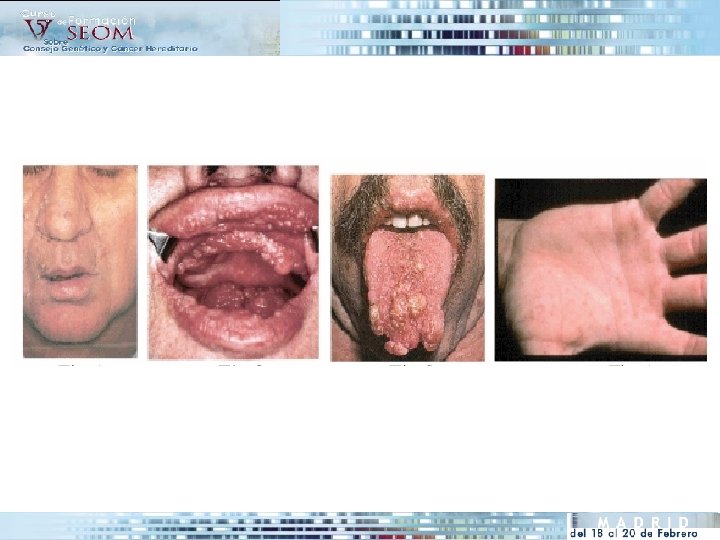
SÍNDROME MUIR-TORRE
• Herencia Autosómica Dominante • Incidencia 2 -3% de todos los casos de cáncer colorrectal • Etiología: mutaciones en genes reparadores del ADN MLH 1, MSH 2, . . ONCOGuia del consejo y asesoramiento genéticos en el cancer hereditario http: //www. aatrm. net/
Sd. de Lynch + ADENOMAS / ADENOCARCINOMA SEBACEOS Y QUERATOACANTOMAS MÚLTIPLES
? ? <50 >70 3 65 GAS 50 ORL 35 52 ESÒFAG PÒLIPS 33 24 31 29 25 50 35 47 45 PÒLIPS 44 ADENOMA SEBACI (NAS) 34 CCR S. MUIR-TORRE? 35 CCR 33 19 14 30 29 26 55 GAS 45 37 ENDO 49 Colono - 8 Gi-CCR 25 23/07/04 31 30 23 Colono - 21 17 Colono - 13
? ? <50 >70 3 65 GAS 50 ORL 35 52 ESÒFAG PÒLIPS 33 24 31 29 25 50 35 47 34 CCR 45 PÒLIPS 44 ADENOMA SEBACI? S. MUIR-TORRE? RER + 35 CCR 33 19 14 30 29 26 55 GAS 45 37 ENDO 49 Colono - 8 Gi-CCR 25 23/07/04 31 30 23 Colono - 21 17 Colono - 13
? ? <50 >70 3 65 GAS 50 ORL 35 52 ESÒFAG PÒLIPS 33 24 31 29 25 50 35 47 45 PÒLIPS 34 CCR 44 ADENOMA SEBACI (NAS) S. MUIR-TORRE? RER + Gi-46 35 CCR 33 19 14 30 29 26 55 GAS 45 37 ENDO 49 Colono - 8 Gi-CCR 25 23/07/04 31 30 23 Colono - 21 17 Colono - 13
? ? <50 >70 3 65 GAS 50 ORL 35 52 ESÒFAG PÒLIPS 33 24 31 29 25 50 35 47 45 PÒLIPS 34 CCR 44 ADENOMA SEBACI (NAS) S. MUIR-TORRE? RER + Gi-46 MSH 2 942+3 A>T 35 CCR 33 19 14 30 29 26 55 GAS 45 37 ENDO 49 Colono - 8 Gi-CCR 25 23/07/04 31 30 23 Colono - 21 17 Colono - 13
? ? <50 >70 3 65 GAS 50 ORL 35 52 ESÒFAG PÒLIPS 33 24 31 29 25 50 35 47 45 PÒLIPS 34 CCR 44 ADENOMA SEBACI (NAS) S. MUIR-TORRE? RER + Gi-46 MSH 2 942+3 A>T 35 CCR 33 19 14 30 29 26 55 GAS 45 37 ENDO 49 Colono - 8 Gi-CCR 25 23/07/04 31 30 23 Colono Gi-166 21 17 Colono Gi-167 13
? ? <50 >70 3 65 GAS 50 ORL 35 52 ESÒFAG PÒLIPS 33 24 31 29 25 50 35 47 45 PÒLIPS 34 CCR 44 ADENOMA SEBACI (NAS) S. MUIR-TORRE? RER + Gi-46 MSH 2 942+3 A>T 35 CCR 33 19 14 30 29 26 55 GAS 45 37 ENDO 49 Colono - 8 Gi-CCR 25 23/07/04 31 30 23 21 17 Colono - Colono Gi-166 Gi-167 MSH 2 WT 13
? ? <50 >70 3 65 GAS 50 ORL 35 52 ESÒFAG PÒLIPS Gi-173 MSH 2 WT 33 24 31 29 25 50 35 47 45 PÒLIPS 34 CCR 44 ADENOMA SEBACI (NAS) S. MUIR-TORRE? RER + Gi-46 MSH 2 942+3 A>T 35 CCR 33 19 14 30 29 26 55 GAS 45 37 ENDO 49 Colono - 8 Gi-CCR 25 23/07/04 31 30 23 21 17 Colono - Colono Gi-166 Gi-167 MSH 2 WT 13
? ? <50 >70 3 65 GAS 50 ORL 52 ESÒFAG PÒLIPS Gi-173 MSH 2 WT 50 55 GAS 47 45 PÒLIPS 34 CCR 44 ADENOMA SEBACI (NAS) S. MUIR-TORRE? 35 CCR 45 37 ENDO 49 Colono Gi-190 RER + Gi-46 MSH 2 942+3 A>T 35 Gi-191 33 Gi-192 24 31 29 Gi-193 25 35 33 19 14 30 29 26 8 Gi-CCR 25 23/07/04 31 30 23 21 17 Colono - Colono Gi-166 Gi-167 MSH 2 WT 13
? ? <50 >70 3 65 GAS 50 ORL 52 ESÒFAG PÒLIPS Gi-173 MSH 2 WT 50 55 GAS 47 49 45 PÒLIPS Colono 34 CCR Gi-190 44 ADENOMA MSH 2 SEBACI (NAS) WT S. MUIR-TORRE? 35 CCR 45 37 ENDO RER + Gi-46 MSH 2 942+3 A>T 35 Gi-191 MSH 2 WT 33 Gi-192 MSH 2 WT 24 31 29 Gi-193 MSH 2 WT 25 35 33 19 14 30 29 26 8 Gi-CCR 25 23/07/04 31 30 23 21 17 Colono - Colono Gi-166 Gi-167 MSH 2 WT 13
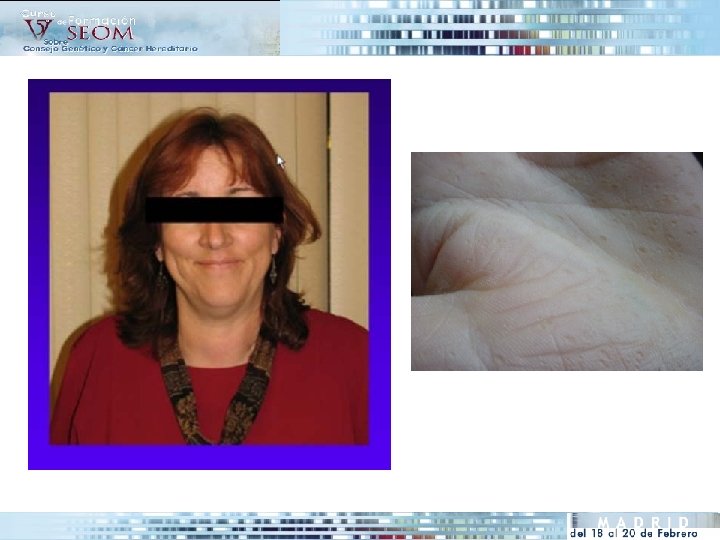
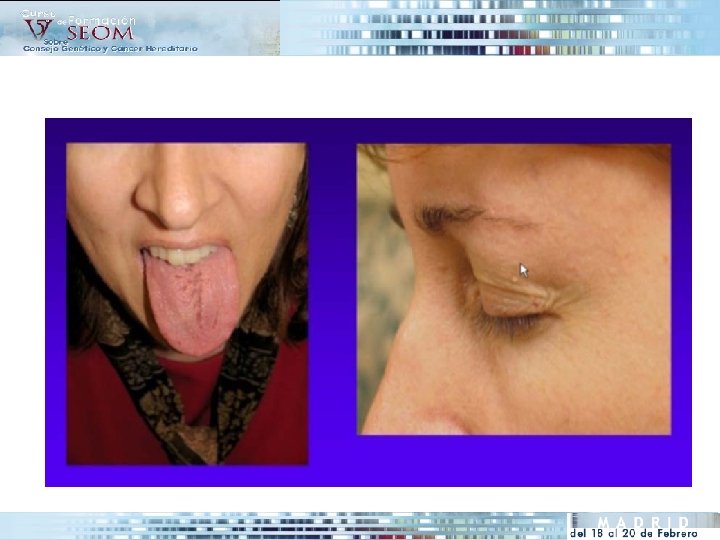
SÍNDROME DE COWDEN PTEN Hamartoma Tumor Syndrome (PHTS)
• Herencia Autosómica Dominante. Casos “de novo” • Incidencia 1/200. 000 -300. 000. Infraestimada • Etiología: mutaciones en el gen PTEN (10 q 23). Tumores asociados a Sd. De Cowden 1. Cáncer de mama (25 -35%) 2. Cáncer de tiroides folicular> papilar 10% 3. Cáncer de endometrio 4. Cáncer renal
60 CEREBRAL >80 73 70 FETGE >70 53 51 >30 >35 >30 28 49 70 >30 25 14 18 14 40 42 43 MAM 44 Tiroides 9 15 20 38 13 14 36 8 5 67 76 32 34 5 2 50 Cirrosis 30 28 3 m 65 Renal 26 8 Gi-MAM 18 19/03/04
60 CEREBRAL 73 70 FETGE >70 53 >30 >80 51 49 LESIONS GENIVES DENTS S. COWDEN >35 >30 28 70 40 42 43 MAM 44 Tiroides 38 36 NO LESIÓ >30 25 14 18 14 9 15 20 13 14 8 5 67 76 32 34 LESIONS CUTANIES ¿SINDROME COWDEN? 5 2 50 Cirrosis 30 28 3 m 65 Renal 26 8 Gi-MAM 18 19/03/04
60 CEREBRAL 73 70 FETGE >70 53 51 49 LESIONS GENIVES S. COWDEN >30 >80 >35 >30 28 70 40 42 43 MAM 44 Tiroides 38 36 NO LESIÓ Gi-242 >30 25 14 18 14 9 15 20 13 14 8 5 67 76 34 LESIONS CUTANIES ¿SINDROME COWDEN? 5 2 32 50 Cirrosis 30 28 3 m 65 Renal 26 8 Gi-MAM 18 19/03/04
60 CEREBRAL 73 70 FETGE >70 53 51 49 LESIONS GENIVES S. COWDEN >30 >80 >35 >30 28 70 >30 25 14 18 14 40 42 43 MAM 44 Tiroides BRCA 1 NO LESIÓ Gi-242 PTEN WT 9 15 20 38 13 14 36 8 5 67 76 32 34 LESIONS CUTANIES ¿SINDROME COWDEN? 5 2 50 Cirrosis 30 28 3 m 65 Renal 26 8 Gi-MAM 18 19/03/04
PROGERIA
• De novo ¿Mosaicismo? • Incidencia 1: 8. 000 • Penetrancia 100% • G 608 G mutation en exon 11 del gen lamina nuclear (LMNA), Manifestaciones clínicas Baja estatura y alteración del crecimiento Facies característica Alopecia Perdida de grasa subcutánea Malformaciones esqueléticas Ateroesclerosis progresiva (exitus por IAM/ICTUS antes de los 15 años)
Mensaje para casa Las enfermedades hereditarias con manifestaciones cutáneas que se asocian a la predisposición a padecer cáncer son entidades heterogéneas y forman parte de síndromes complejos con afectación multisistémica Las manifestaciones cutáneas son criterios diagnósticos mayores y/o patognomónicos con frecuencia en estos síndromes (e. g. Sd. Cowden, Sd Gorlin, Sd. Birt-Hogg-Dubè ) El examen dermatológico debe estar integrado en la valoración de estos pacientes El asesoramiento genético en estas familias permite identificar a individuos en riesgo y establecer medidas de cribado y preventivas así como en casos seleccionados establecer estrategias reproductivas