Talasemias Talasemia La talasemia es un grupo heterogneo




, siendo")
















. • La Hb. E es el resultado de la sustitución")
. En la Hb Lepore existe un entrecruzamiento no homologo (crossing-over)")


. Radiografía de cráneo (cráneo en cepillo). Electroforesis de hemoglobinas,")





- Slides: 32
Talasemias
Talasemia La talasemia es un grupo heterogéneo de defectos congénitos de la hemoglobina que se transmiten de forma autosómica recesiva con expresividad variable(síndromes talasémicos), cuya consecuencia es la disminución o ausencia de la síntesis de cadenas de globina estructuralmente normales.
Historia Thomas Cooley Esta entidad clínica fue descrita por primera vez por los médicos Thomas Cooley y Pearl Lee de Chicago en 1925 y detectada por primera vez en un niño inmigrante italiano.
Etiología El nombre de esta condición se deriva del griego Thalassa, mar y haema, sangre. Esto es debido a que los primeros casos se observaron el individuos cuyos antepasados eran originarios de las tierras que bordean el Mar Mediterraneo. El término fue utilizado por primera vez en 1932.
Epidemiología La distribución mundial de las talasemias, coincide con la del paludismo (Malaria), siendo muy frecuente en los países de la Cuenca del Mediterráneo, Norte de África, Oriente, India y Sudeste Asiático. La migración de las poblaciones ha extendido a las talasemias a otros países europeos, América del Norte, Sudamérica y Australia. Boletín de la Organización Mundial de la Salud Recopilación de artículos Volumen 86: 2008 Volumen 86, junio 2008, 419 -496 http: //www. who. int/bulletin/volumes/86/6/06 -036673 -ab/es/
Epidemiología cont… Cada año nacen más de 330 000 niños afectados con una hemoglobinopatía donde el 17% de casos se debe a talasemia. Las hemoglobinopatías causan aproximadamente un 3, 4% de las defunciones entre los niños menores de 5 años. A nivel mundial, en torno a un 7% de las mujeres embarazadas son portadoras de talasemia β o α cero, o de hemoglobina S, C, D Punjab o E. Boletín de la Organización Mundial de la Salud Recopilación de artículos Volumen 86: 2008 Volumen 86, junio 2008, 419 -496 http: //www. who. int/bulletin/volumes/86/6/06 -036673 -ab/es/
Clasificación Alfa Beta Delta - Beta
Gravedad clínica Los términos para indicar la gravedad clínica de una talasemia son los siguientes: TALASEMIA MAYOR TALASEMIA INTERMEDIA TALASEMIA MENOR (MÍNIMA)
Mecanismo Fisiopatológico de las Manifestaciones Exceso de cadenas libres Precipitados Eritrocito Hemolisis Eritropoyetin a Expansión de la M. O Alteraciones del esqueleto Lesión de la membrana celular Anemia Absorción de hierro Medula ósea Transfusiones Sobrecarga de hierro Hemocromatosis secundaria
α- Talasemia
α 0 - talasemia o alfatalasemia tipo I Obedece a la deleción de 2 genes alfa y, en su forma heterocigota, presenta una expresividad clínica superponible a la de betatalasemia menor. Su forma homocigota corresponde a la hidropesía fetal
α 0 -talasemia con Hb Barts Esta forma de alfatalasemia por deleción de todos los genes de alfaglobina es incompatible con la vida. En general, constituye una causa de aborto hacia las 30 semanas del embarazo o de muerte fetal poco después del nacimiento por anasarca fetoplacentaria (hidropesía fetal). Manifestaciones: Niño pálido edematoso Insuficiencia cardíaca Anemia prolongada intrauterina Hepatoesplenomegalia pronunciada Retraso en el crecimiento del cerebro Deformidades esqueléticas y cardiovasculares. Aumento brusco de la placenta. Suelen morir en el útero (23 -28 semanas) o al nacer.
α +-talasemia o alfatalasemia tipo 2 Obedece a la deleción de un único gen alfa. En su forma heterocigota constituye una forma de talasemia mínima. Su carácter asintomático pasa prácticamente siempre desapercibida.
Hemoglobinopatía H Presenta una expresividad clínica superponible a la betatalasemia intermedia con signos de hemólisis crónica y esplenomegalia. Su presencia se ha observado también asociada a retraso mental o de forma adquirida en el curso de mielodisplasias y leucemias.
β-Talasemia
β - Talasemia menor Es la forma más frecuente de talasemia Se caracteriza por una seudopoliglobulia microcítica con anemia muy discreta o inexistente. Se presencia hipocromía Rara vez se aprecia esplenomegalia.
β - Talasemia intermedia El cuadro clínico es siempre manifiesto y se caracteriza por: Anemia de intensidad moderada Hemólisis crónica , Esplenomegalia, cuya gravedad no alcanza nunca la de la Enfermedad de Cooley.
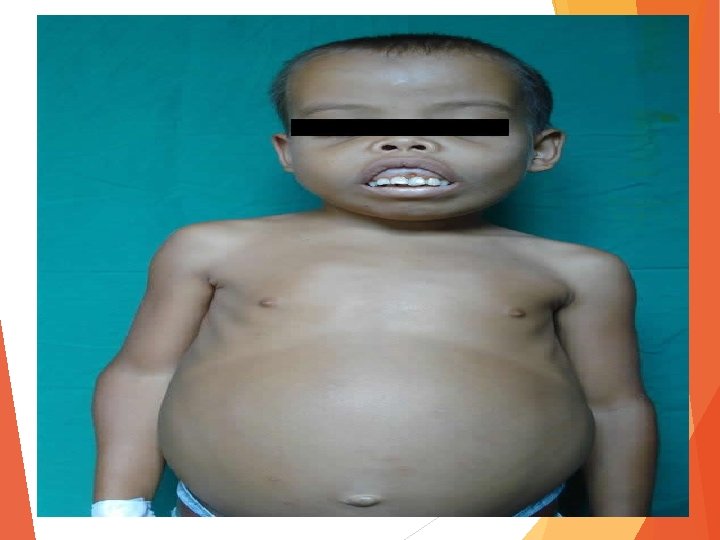
Β - TALASEMIA MAYOR O ANEMIA DE COOLEY Esta se inicia a partir de los 6 meses del nacimiento y se caracteriza por: • Intensa anemia • Esplenomegalia, a veces gigante • Hepatomegalia.
El estudio radiológico muestra la imagen del llamado “cráneo en cepillo”.
DELTA- BETA TALASEMIA • La delta-beta-talasemia es una forma de BT caracterizada por una disminución o ausencia de síntesis de las cadenas de delta y beta globina con un aumento compensador de la síntesis de las cadenas gamma. • Se desconoce la prevalencia aunque, es muy frecuente en Grecia, Italia y España.
HEMOGLOBINOPATÍAS TALASÉMICAS
Hemoglobina E (Hb. E). • La Hb. E es el resultado de la sustitución del acido glutámico (Glu) en posición 26 por la lisina (Lys) en la cadena beta. • Esta mutación activa una región de escisión (splicing) criptica que se manifiesta por un defecto en la maduración del RNAm y disminución de la síntesis de cadenas de betaglobina.
Hemoglobina Lepore (Hb Lepore). En la Hb Lepore existe un entrecruzamiento no homologo (crossing-over) con intercambio de material genético y formación de un gen hibrido 5 beta. Este gen codifica una cadena globinica hibrida cuya secuencia esta constituida por un fragmento (50 -80 aminoácidos) de cadena 5 y un fragmento (60 a 90 aminoácidos) de cadena beta, ambos normales.
Alargamiento de la cadena normal de globina Las mutaciones con alargamiento de una cadena normal de globina obedecen en su mayoria a la desaparicion del codon de terminacion (UAA, UAG, UGA). Debido a ello, en el proceso de traduccion, la cadena de globina continua hasta hallar otro codon terminal, lo que supone un alargamiento de la cadena. Un ejemplo de mutacion del codon terminal en la region a es la Hb Constant Spring (Hb. CS), y en la region (3, la Hb. Tak.
Síntesis de cadenas globínicas muy inestables. Algunas mutaciones de los genes a o [3 tienen como consecuencia un defecto en la traducción del RNAm, por el cual se produce la síntesis de cadenas globínicas muy inestables. En la mayoría de este tipo de variantes el nivel de RNAm alterado es normal, pero las cadenas de globina, resultado de su traducción, son muy inestables y se degradan con gran rapidez, a no ser que sean incorporadas inmediatamente al tetrámero de Hb, con lo que adquieren mayor estabilidad.
Diagnostico Hemograma (anemia microcítica hipocrómica). Radiografía de cráneo (cráneo en cepillo). Electroforesis de hemoglobinas, donde se observa un aumento característico de la fracción Hb. A 2 (3, 8 -7%) con Hb. F normal (< 2%).
Diagnostico Diferencial RDW Fe sérico TIBC Ferritina sérica FEP Nivel A 2 Deficiencia de hierro N -talasemia N N N N N Enfermedad de Hb. E N N N Anemia de enf. crónica N N Anemia sideroblástica N N Intoxicación por plomo N N N
Diagnostico Definitivo: Pruebas de Mecanismo moleculares • Consiste en la incubación de DNA mediante enzimas de restricción, que lo seccionan selectivamente a nivel de determinados pares de bases, dando lugar a diferentes fragmentos La técnica de llamados «de restricción» Southern-blot Reacción en cadena de la polimerasa o PCR • Es un método rápido, sencillo y altamente sensible, que permite amplificar secuencias específicas de DNA o RNA mediante la obtención de un elevado número de copias de las mismas.
TRATAMIENTO El tratamiento será determinado por su médico basándose en lo siguiente: Su edad, su estado general de salud y su historia médica. Qué tan avanzada está la enfermedad Su tolerancia a determinados medicamentos, procedimientos o terapias. Sus expectativas para la trayectoria de la enfermedad. Su opinión o preferencia.
TRATAMIENTO Puede incluir: Dosis diarias de ácido fólico. Ningún suplemento de hierro. Transfusiones de sangre regulares. Medicamentos (para disminuir la cantidad de hierro en el cuerpo, llamada terapia de quelación). Esplenectomía (extirpación quirúrgica del bazo). Posible extirpación quirúrgica de la vesícula biliar. Trasplante de médula ósea