Sympatholytic drugs Adrenergic blockers BY NOHAD A ATRUSHI

Sympatholytic drugs: α- Adrenergic blockers BY NOHAD A ATRUSHI 23/11/2014
 Dibenamine Phenoxybenzamine*")
Alpha- Adrenergic Antagonists I. Non-selective alpha adrenergic receptor antagonists A. Covalent (haloalkylamines) Dibenamine Phenoxybenzamine* B. Noncovalent Phentolamine* Tolazoline II. a 1 -selective Doxazosin Prazosin* Terazosin III. a 2 -selective Yohimbine*

Structure of several a -receptor-blocking drugs.
 α-Adrenergic Blocking Agents �The α-adrenergic blocking")
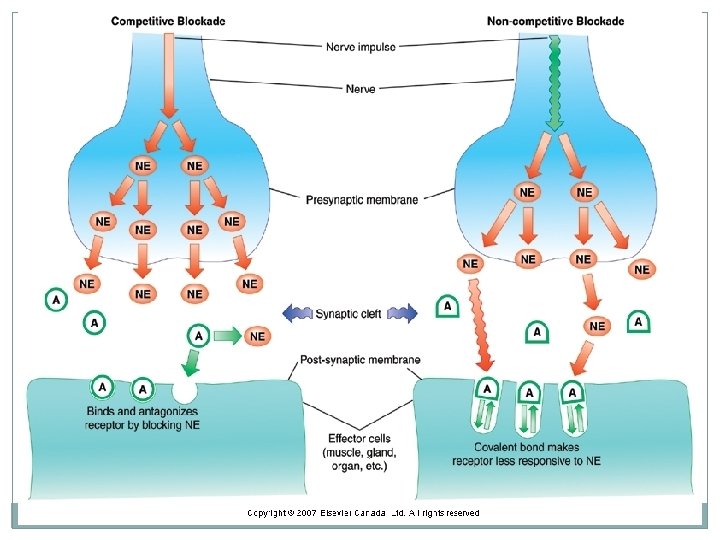
Adrenergic Antagonists (also called blockers or sympatholytic agents) α-Adrenergic Blocking Agents �The α-adrenergic blocking agents, phenoxybenzamine and phentolamine, have limited clinical applications, they are nonselective α blockers

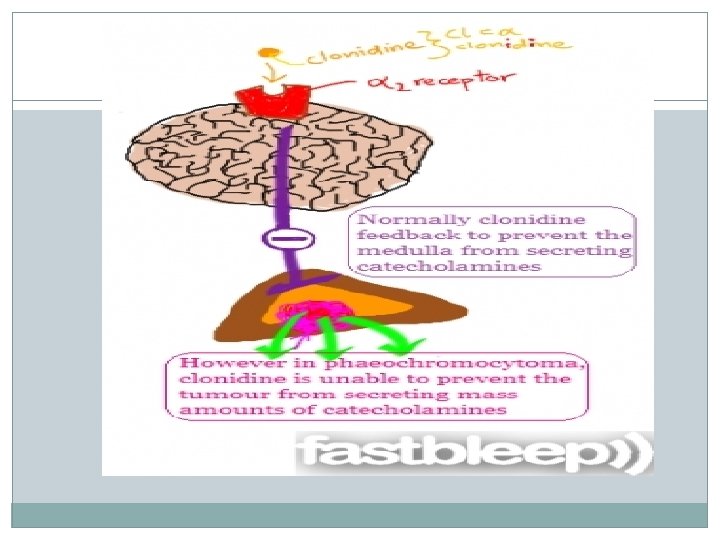
1. Beta-haloalkylamine Phenoxybenzamine, Dibenamine • is used in the treatment of pheochromocytoma, a catecholamine-secreting tumor of the adrenal medulla. • Adverse effects: Phenoxybenzamine can cause postural hypotension, nasal stuffiness, nausea, and vomiting.

Selective a 1 blockers cause less reflex tachycardia than Phenoxybenzamine and Phentolamine

Phenoxybenzamine � Non specific, long acting irreversible alpha antagonist � MOA: Spontaneously cyclizes in the body to give ethyleniminium intermediate – forms a strong covalent bond with α receptors – blockade of alpha receptor (lasts for 3 – 4 days) � Dibenamine

Covalent inactivation of a-receptor by phenoxyben CH 2 O CH 2 N CH 2 Cl CH Phenoxybenzamine CH 3 CH 2 + N CH CH 2 CH 3 Ethylene iminium ion O CH 2 N CH CH 2 CH 3 Alkylated a-receptor

�Phenoxybenzamine forms a permanent covalent bond with adrenergic receptors. Based on known information about the structures of these receptors, it likely involves attack by the cysteine at position 3. 36 in transmembrane helix 3 to form a stable linkage. Thus, it remains permanently bound to the receptor, preventing adrenaline and noradrenaline from binding. This causes vasodilatation in blood vessels, due to its antagonistic effect at the alpha-1 adrenoceptor found in the walls of blood vessels, resulting in a drop in blood pressure. A side effect of phenoxybenzamine is reflex tachycardia.

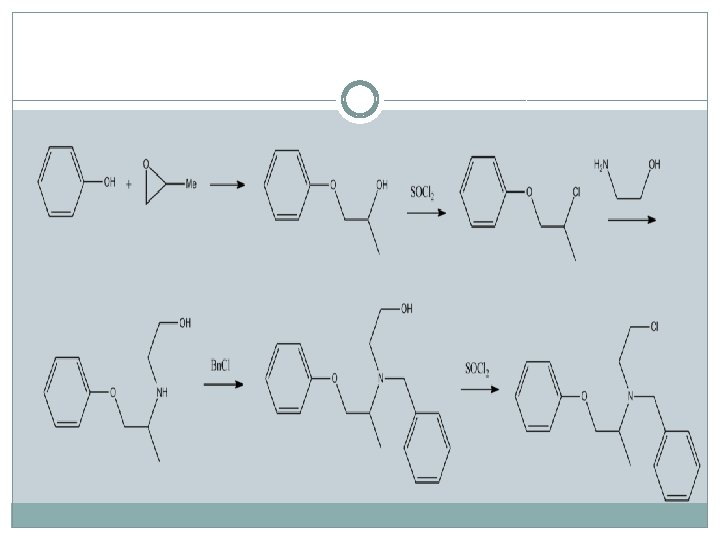
�Phenoxybenzamine can be synthesized by reacting phenol with propylenoxide, which forms 1 phenoxy-2 -propanol, the chlorination of which with thionyl chloride gives 1 -phenoxy-2 propylchloride. Reacting this with 2 -aminoethanol leads to formation of 1 -phenoxy-2 -(2 hydroxyethyl)aminopropane. Alkylation of the secondary amino group gives N-(2 -hydroxyethyl)-N(1 -methyl-2 -phenoxyethyl)benzylamine, the hydroxyl group of which is chlorinated using thionyl chloride, giving phenoxybenzamine.

2. Phentolamine �Phentolamine is also used for the short-term management of pheochromocytoma. �Non specific, short acting reversible alpha antagonist �Potent competitive antagonist at both 1 and 2 receptors �Quick acting (in minutes) �Reduction in Peripheral Resistance - blocking both α -1 and α-2 receptors - causes NA release and venodilatation more than arteriolar
phenol using 2 chloromethylimidazoline")
�Phentolamine can be synthesized by alkylation of 3 - (4 -methylanilino)phenol using 2 chloromethylimidazoline

�Prazosin, the first known selective α 1 -blocker, was discovered in the late 1960 s , and is now one of a small group of selective α 1 -antagonists that includes three other quinoxaline antihypertensives terazosin, doxazosin, and alfuzosin and the nonquinazoline benzensulfonamides tamsulosin and silodosin . Prazosin, doxazosin, and terazosin contain a 4 -amino 6, 7 -dimethoxyquinazoline ring system attached to a piperazine ring, whereas alfuzosin has a rotatable propylenediamine group (an open piperazine ring). The other structural differences are the heterocyclic acyl groups attached to the second nitrogen of the piperazine or the propyl chain.

�The differences in these groups afford dramatic differences in some of the pharmacokinetic properties of these agents. For example, reduction of the furan ring for prazosin to the tetrahydrofuran ring of terazosin increases its duration of action by altering its rate of metabolism. Some of the important clinical parameters of the quinazolines. The long half-lives and durations of action for terazosin, doxazosin, tamsulosin, and silodosin permit once-a-day dosing and generally lead to increased patient compliance.
 � Non-specific blockade of all subtypes")
Prazosin � Highly selective alpha-1 blocker (1: 1000) � Non-specific blockade of all subtypes - α 1 A, α 1 B and α 1 D � Blockade of sympathetic vasoconstriction - fall in BP � NA is not released as α-2 is not blocked (only mild tachycardia) � Dilates arterioles more than veins – Postural hypotension is less – only 1 st dose effect (dizziness and fainting) � Kinetics: effective orally (70%), metabolized in liver and half life is 6 -8 Hrs � Uses: Hypertension Raynaud`s disease BHP

Disorders of the Autonomic Nervous System: Raynaud’s Disease � Raynaud’s disease – characterized by constriction of blood vessels Provoked by exposure to cold or by emotional stress

3. Prazosin terazosin, doxazosin, and tamsulosin • are selective competitive blockers of the α 1 receptor. • The first three drugs are useful in the treatment of hypertension. • Tamsulosin is indicated for the treatment of benign prostatic hyperplasia. • Doxazosin is the longest acting of these drugs.

�The first dose of these drugs produces an exaggerated orthostatic hypotensive response that can result in syncope (fainting). This action, termed first-dose effect. �Tamsulosin is a more potent inhibitor of the α 1 A receptors found on the smooth muscle of the prostate. This selectivity accounts for tamsulosin's minimal effect on blood pressure.

4. Yohimbine �Is a selective α 2 blocker. �It is found as a component of the bark of the yohimbe tree and is sometimes used as a sexual stimulant (aphrodisiac) or cardiovascular stimulant.

Comparison of alpha blockers Receptor affinity


Sympatholytic drugs: beta- adrenergic blockers BY NOHAD A ATRUSHI 26/11/2014

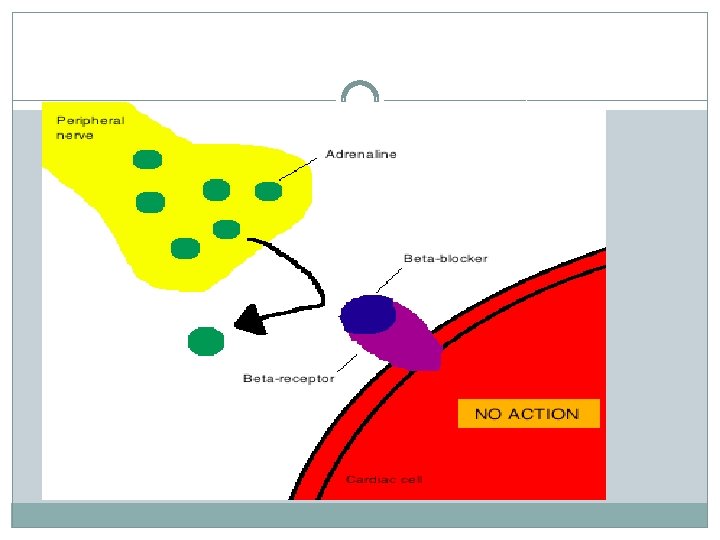
β-Adrenergic Blocking Agents � Nonselective β-blockers act at both β 1 and β 2 receptors, whereas cardioselective β antagonists block β 1 receptors �[Note: There are no clinically useful β 2 blockers]. � Although all β-blockers lower blood pressure in hypertension, they do not induce postural hypotension, because the α-adrenoceptors remain functional.

β-Blockers are also effective in treating angina, cardiac arrhythmias, myocardial infarction, congestive heart failure, hyperthyroidism, glaucoma, as well as serving in the prophylaxis of migraine headaches.

Structures of some b-receptor antagonists. 0
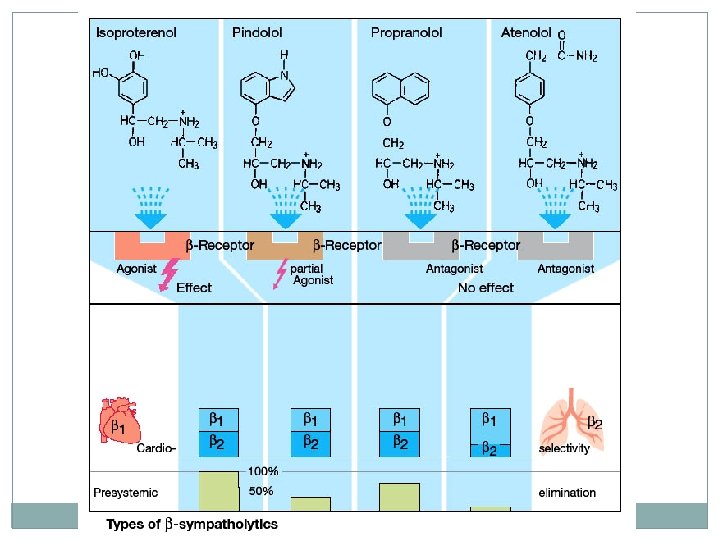

1. Propranolol �A nonselective β blocker • Sustained-release preparations for once-a-day dosing are available. • Actions: • Cardiovascular: Propranolol diminishes cardiac output, having both negative inotropic and chronotropic effects. • Cardiac output, work, and oxygen consumption are decreased by blockade of β 1 receptors; these effects are useful in the treatment of angina. • The reduction in cardiac output leads to decreased blood pressure.

�Bronchoconstriction: Blocking β 2 receptors in the lungs of susceptible patients causes contraction of the bronchiolar smooth muscle. �Non-selective β-blockers, are contraindicated in patients with COPD or asthma. �β-blockade leads to decreased glycogenolysis and decreased glucagon secretion, thus pronounced hypoglycemia may occur after insulin injection in a patient using propranolol. � β-Blockers also mask the normal physiologic response to hypoglycemia.

Mechanisms of action �Propranolol lowers blood pressure in hypertension by: Decreased cardiac output is the primary mechanism, inhibition of renin release from the kidney and decreased sympathetic outflow from the CNS also contribute to propranolol's antihypertensive effects.

�Adverse effects: Bronchoconstriction Arrhythmias: Treatment with β-blockers must never be stopped quickly because of the risk of precipitating cardiac arrhythmias, which may be severe. Sexual impairment

�Drug interactions: Drugs that interfere with the metabolism of propranolol, such as cimetidine, fluoxetine, paroxetine, and ritonavir, may potentiate its antihypertensive effects. Conversely, those that stimulate its metabolism, such as barbiturates, phenytoin, and rifampin, can decrease its effects.

2. Timolol and nadolol: • Nonselective β blockers, • are more potent than propranolol. 3. Acebutolol, atenolol, metoprolol, and esmolol: • Selective β 1 blockers • Esmolol has a very short lifetime. It is only given intravenously if required during surgery or management of poisoning. 4. Pindolol and acebutolol: • blockers with partial agonist activity

5. Labetalol and carvedilol: • blockers of both α- and β- adrenoceptors • Carvedilol also decreases lipid peroxidation and vascular wall thickening, effects that have benefit in heart failure. • Labetalol may be employed as an alternative to methyldopa in the treatment of pregnancy-induced hypertension. • Intravenous labetalol is also used to treat hypertensive emergencies, because it can rapidly lower blood pressure.

Drugs Affecting Neurotransmitter Release or Uptake • Some agents act on the adrenergic neuron, either to interfere with neurotransmitter release or to alter the uptake of the neurotransmitter. 1. Reserpine • Reserpine, a plant alkaloid that causes the depletion of biogenic amines. • Sympathetic function, in general, is impaired because of decreased release of norepinephrine.

2. Guanethidine • Guanethidine blocks the release of stored norepinephrine as well as displaces norepinephrine from storage vesicles (thus producing a transient increase in blood pressure). • This leads to gradual depletion of norepinephrine in nerve endings except for those in the CNS. • Guanethidine commonly causes orthostatic hypotension and interferes with male sexual function.

3. Alpha methyl dopa �Antihypertensive �Mechanism: Transformed to alpha methyl noradrenaline in adrenergic neuron, �When released, alpha methyl noradrenaline acts as agonist on presynaptic alpha 2 receptors. Thus further release of transmitter is inhibited.
 �β-adrenergic receptor antagonists (beta-blockers/β- blockers) were initially developed in the")
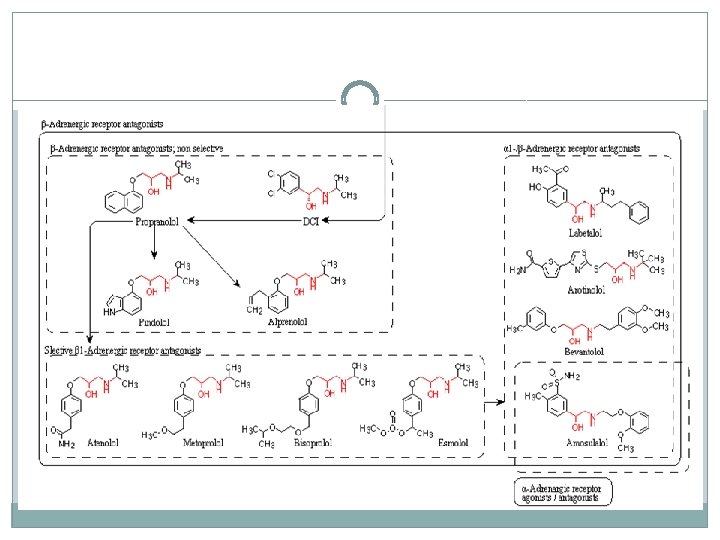
beta-adrenergic receptor antagonists (betablockers) �β-adrenergic receptor antagonists (beta-blockers/β- blockers) were initially developed in the 1960 s, for the treatment of angina pectoris but are now also used for hypertension, congestive heart failure and certain arrhythmias. [1] In 1950 s, dichloroisoproterenol (D CI) was discovered to be a β-antagonist that blocked the effects of sympathomimetic amines on bronchodilation, uterine relaxation and heart stimulation. Although DCI had no clinical utility, a change in the compound did provide a clinical candidate, pronethalol, which was introduced in 1962.

The evolution of non-selective and selective β-blockers �By the time propranolol was launched, ICI was beginning to experience competition from other companies. This potential threat led to ongoing refinements in the pharmacologic structure of βblockers and subsequent advances in drug delivery. ICI studied analogues further and in 1970 launched practolol, under the trade name Eraldin®. It was withdrawn from the market a few years later because of the severe side effects it caused, nevertheless it played a large role in the fundamental study of β-blockade and β-receptors.

�The withdrawal of Eraldin® gave ICI the nudge to launch another β-blocker, atenolol, which was launched in 1976 under the trade name Tenormin®. Atenolol is a selective β 1 -receptor antagonist and was developed for the purpose of obtaining the “ideal βblocker”. It soon became one of the best-selling heart drug. ICI’s β-blocker project was based on Ahlquist’s dual receptor theory. The drugs that were the outcome of this project, from propranolol to atenolol, helped to establish the receptor theory among scientist and pharmaceutical companies.

�The progress in β-blocker development led to the introduction of drugs with variety of properties. βblockers were developed having a relative selectivity for cardiac β 1 -receptors (for example metoprolol and atenolol), partial adrenergic agonist activity (pindolol), concomitant α-adrenergic blocking activity (for example labetalol and carvedilol) and additional direct vasodilator activity (nebivolol). In addition, longacting and ultra-short formulations of β-blockers were developed. [7] In 1988, Sir James Black was awarded the Nobel Prize in Medicine for his work on drug development. [3][4][6][7]

Binding to β-adrenergic receptors �Figure 3: β-blockers cause a competitive inhibition of the β-receptor, which counters the effects of catecholamines. �Three different types of β-adrenergic receptors have been identified by molecular pharmacology. β 1 -receptorsare located in the heart and consist of about 75% of all βreceptors. β 2 -receptors can be found in the smooth muscles of vessels and the bronchies. β 3 -receptors are presumed to be involved in fatty acid metabolism and are found in the adipocytes. [9] β-blockers cause a competitive inhibition of the β-receptor, which counters the effects of catecholamines. �

�β 1 and β 2 -receptors are G-protein coupled receptors, which couple to Gαs-proteins. When activated, it stimulates an increase in intracellular c. AMP via the adenylyl cyclase. c. AMP, which is the second messenger, then activates protein kinase A that phosphorylates the membrane's calcium channeland increases the entry of calcium into the cytosol. Protein kinase A also increases the release of calcium from the sarcoplasmic reticulum, which causes a positive inotropic effect. The phosphorylation of troponin I andphospholamban by protein kinase A causes the lusitropic effects of βblockers. This increases the re-uptake of calcium by the sarcoplasmic reticulum.

�β-blockers are sympatholytic drugs. Some β-blockers partially activate the receptor while preventing catecholamines from binding to the receptor, making them partial agonists. They provide a background of sympathetic activity, while preventing normal and enhanced sympathetic activity. These β-blockers possessintrinsic sympathomimetic activity (ISA). Some of them also possess what is called membranestabilizing activity(MSA) on myocardial muscle fibers.

A few of the non-selective β-blockers Labetalol Pindolol Propranolol Timolol

A few of the selective β 1 -blockers Atenolol Acebutolol Bisoprolol Nebivolol Metoprolol

Selectivity �β-blockers can be selective for either β 1 or β 2 - receptor or non-selective. By blocking β 1 -receptor it is possible to reduce heart rate, conduction of velocity and contractility. The blocking of β 2 receptor promotes vascular smooth muscle contraction, which results in increase of peripheral resistance. Blockade of the β 2 -receptor effectively reduces the sympathetic activity, which results in reduce of the associated platelet and coagulation activation. This is why a non-selective β-blocker treatment may result in a lower risk of both arterial and venous embolic events.
![(S)-propranolol[edit] �Propranolol exist in two different enantiomers, (S)- and (R)-enantiomers. The (S)-isomer is 100](http://slidetodoc.com/presentation_image_h/27aa0bd58654a13edb37ff713291ae0f/image-52.jpg "(S)-propranolol[edit] �Propranolol exist in two different enantiomers, (S)- and (R)-enantiomers. The (S)-isomer is 100")
(S)-propranolol[edit] �Propranolol exist in two different enantiomers, (S)- and (R)-enantiomers. The (S)-isomer is 100 -fold more potent than the (R)-isomer and that is the general rule for most β-blockers. It is possible to produce the (S)propranolol enantiomer from α-naphthol and 3 bromopropanol as seen in figure 6. � α-Naphthol and 3 -bromopropanol are refluxed for 6 hours to give alcohol. The alcohol is oxidized by using 2 Iodoxybenzoic acid (IBX) to givealdehyde. The aldehyde is subjected to L-prolinecatalyzed asymmetric αaminoxylation and a reduction is made with Na. BH 4 in methanol. A diol is obtained by Pd/Ccatalyzed hydrogenolysis. Finally the diol is converted to epoxide using the Mitsunobu reaction and stirred with isopropyl amine in CH 2 Cl 2 to give (S)-propranolol.
-propranolol from αnaphthol and 3 -bromopropanol.")
Figure 6: Synthesis of (S)-propranolol from αnaphthol and 3 -bromopropanol.

Figure 7: The oxymethylene bridge of propranolol can be seen inside the green ring.
 �β-blockers' binding site to the receptor is the same as")
Structure activity relationship (SAR) �β-blockers' binding site to the receptor is the same as for endogenous catecholamines, such as noradrenaline and adrenaline. This binding is based on hydrogen bondsbetween the β-blocker and the receptor, and therefore not based on covalent bonds, which results in the reversibility of the binding. A significant step in the development of βadrenergic antagonists was the discovery that an oxymethylene bridge (-OCH 2) could be inserted into the arylethanolamine structure of pronethalol to produce propranolol.

�Propranolol is an aryloxypropanolamine, which are more potent β-blockers than arylethanolamines. Today, most of the β-blockers used clinically are aryloxypropanolamines. The length of the side chain is increased when an oxymethylene bridge is introduced. It has been shown that the side chains of aryloxypropanolamine can adopt a conformation that puts the hydroxyl and amine groups in more or less the same position as with beta blocker that do not have this group as a part of the side chain. [2]

�After the release of propranolol, relative lipophilicity of β -blockers as a significant factor in their varied and complex pharmacology, became an important factor. It was suspected that propranolol’s centrally induced side effects could be due to its high lipophilicity. Thus, it was focused on synthesizing analogues with hydrophilic moieties, favourably placed to see if the side effects would decrease. �Selecting para-acylamino groups as the hydrophilic moiety, scientists synthesized a group of para-acylphenoxyethanol-and propanolamines, and selected practolol for clinical trials.

�Practolol had one property not previously seen with β-blockers, it exhibited cardioselectivity (β 1 selectivity). Studies from practolol showed that moving the acylamino group to meta or ortho positions, on the benzene ring, caused a loss of selectivity but not loss of the β-blockade itself. This illustrated the significance ofpara-substitution for β 1 selectivity of β-blockers. [5]
 for β-blockers. For the function of")
Figure 8 shows the structural activity relationship (SAR) for β-blockers. For the function of a β-blocker it's essential for the compound to contain an aromatic ring and a βethanolamine. The aromatic ring can either be benzoheterocyclic (such as indol) or heterocyclic (such as thiadiazole). This is mandatory. ] The side chains can be variable. Figure 8: Structural activity relationship for β-blockers. The X part of the side chain can either be directly linked to the aromatic ring or linked through a -OCH 2 - group.

When X is -CH 2 -, -CH=CH-, -SCH 2 - or -NCH 2 - there is little or no activity. The R group can only be a secondary substitution and branched is the optimal choice. Alkyl (CH 3) substituents on the α, β or γ carbon (if X = OCH 2) lower beta blockade, especially at the α carbon. The general rule for aromatic substitution is: ortho > meta > para. This gives non-selective β-blockers. Large para-substituents usually decrease activity but large ortho-groups retain some activity. Polysubstitution on carbon 2 and 6 makes the compound inactive but when the substitution is on carbon 3 and 5 there's some activity. For the highest cardioselectivity, the substituents should be as following: para > meta > ortho. All the β-blockade is in one isomer, (S)-aryloxypropylamine and (R)ethanolamine. [5]
- Slides: 61