Supervised Classification Selection bias in gene extraction on

Supervised Classification

Selection bias in gene extraction on the basis of microarray geneexpression data Ambroise and Mc. Lachlan Proceedings of the National Academy of Sciences Vol. 99, Issue 10, 6562 -6566, May 14, 2002 http: //www. pnas. org/cgi/content/full/99/10 /6562

Supervised Classification of Tissue Samples We OBSERVE the CLASS LABELS z 1, …, zn where zj = i if jth tissue sample comes from the ith class (i=1, …, g). AIM: TO CONSTRUCT A CLASSIFIER c(y) FOR PREDICTING THE UNKNOWN CLASS LABEL z OF A TISSUE SAMPLE y. e. g. g = 2 classes C 1 - DISEASE-FREE C 2 - METASTASES

Sample 1 Sample 2 Expression Signature Gene 1 Gene 2 Expression Profile Gene N Sample M
 Sample 1 . . . . Sample n . .")
Supervised Classification (Two Classes) Sample 1 . . . . Sample n . . . . Gene 1 Gene p Class 1 (good prognosis) Class 2 (poor prognosis)

Microarray to be used as routine clinical screen by C. M. Schubert Nature Medicine 9, 9, 2003. The Netherlands Cancer Institute in Amsterdam is to become the first institution in the world to use microarray techniques for the routine prognostic screening of cancer patients. Aiming for a June 2003 start date, the center will use a panoply of 70 genes to assess the tumor profile of breast cancer patients and to determine which women will receive adjuvant treatment after surgery.
")
Selection Bias that occurs when a subset of the variables is selected (dimension reduction) in some “optimal” way, and then the predictive capability of this subset is assessed in the usual way; i. e. using an ordinary measure for a set of variables.
 Regression: Breiman")
Selection Bias Discriminant Analysis: Mc. Lachlan (1992 & 2004, Wiley, Chapter 12) Regression: Breiman (1992, JASA) “This usage (i. e. use of residual of SS’s etc. ) has long been a quiet scandal in the statistical community. ”

Nature Reviews Cancer, Feb. 2005

LINEAR CLASSIFIER FORM for the production of the group label z of a future entity with feature vector y.

FISHER’S LINEAR DISCRIMINANT FUNCTION where and S are the sample means and pooled sample covariance matrix found from the training data

Microarrays also to be used in the prediction of breast cancer by Mike West (Duke University) and the Koo Foundation Sun Yat-Sen Cancer Centre, Taipei Huang et al. (2003, The Lancet, Gene expression predictors of breast cancer).

LINEAR CLASSIFIER FORM for the production of the group label z of a future entity with feature vector y.

FISHER’S LINEAR DISCRIMINANT FUNCTION where and S are the sample means and pooled sample covariance matrix found from the training data
 where β 0 and β are obtained as follows:")
SUPPORT VECTOR CLASSIFIER Vapnik (1995) where β 0 and β are obtained as follows: subject to relate to the slack variables separable case

with non-zero only for those observations j for which the constraints are exactly met (the support vectors).
 REPLACE by where the kernel function is the inner product")
Support Vector Machine (SVM) REPLACE by where the kernel function is the inner product in the transformed feature space.
 The Lagrange (primal function) is which we maximize")
HASTIE et al. (2001, Chapter 12) The Lagrange (primal function) is which we maximize w. r. t. β, β 0, and ξj. Setting the respective derivatives to zero, we get with and
 to (4) into (1), we obtain the Lagrangian dual function We")
By substituting (2) to (4) into (1), we obtain the Lagrangian dual function We maximize (5) subject to In addition to (2) to (4), the constraints include Together these equations (2) to (8) uniquely characterize the solution to the primal and dual problem.
 Statistical modeling: the two cultures (with discussion). Statistical Science 16, 199")
Leo Breiman (2001) Statistical modeling: the two cultures (with discussion). Statistical Science 16, 199 -231. Discussants include Brad Efron and David Cox
 LEUKAEMIA DATA: Only 2 genes are")
GUYON, WESTON, BARNHILL & VAPNIK (2002, Machine Learning) LEUKAEMIA DATA: Only 2 genes are needed to obtain a zero CVE (cross-validated error rate) COLON DATA: Using only 4 genes, CVE is 2%

Since p>>n, consideration given to selection of suitable genes SVM: FORWARD or BACKWARD (in terms of magnitude of weight βi) RECURSIVE FEATURE ELIMINATION (RFE) FISHER: FORWARD ONLY (in terms of CVE)
 LEUKAEMIA DATA: Only 2 genes are needed to obtain a")
GUYON et al. (2002) LEUKAEMIA DATA: Only 2 genes are needed to obtain a zero CVE (cross-validated error rate) COLON DATA: Using only 4 genes, CVE is 2%
 “The success of the RFE indicates that RFE has a")
GUYON et al. (2002) “The success of the RFE indicates that RFE has a built in regularization mechanism that we do not understand yet that prevents overfitting the training data in its selection of gene subsets. ”
 n=62 (40 tumours; 22")
Example: Microarray Data Colon Data of Alon et al. (1999) n=62 (40 tumours; 22 normals) tissue samples of p=2, 000 genes in a 2, 000 62 matrix.
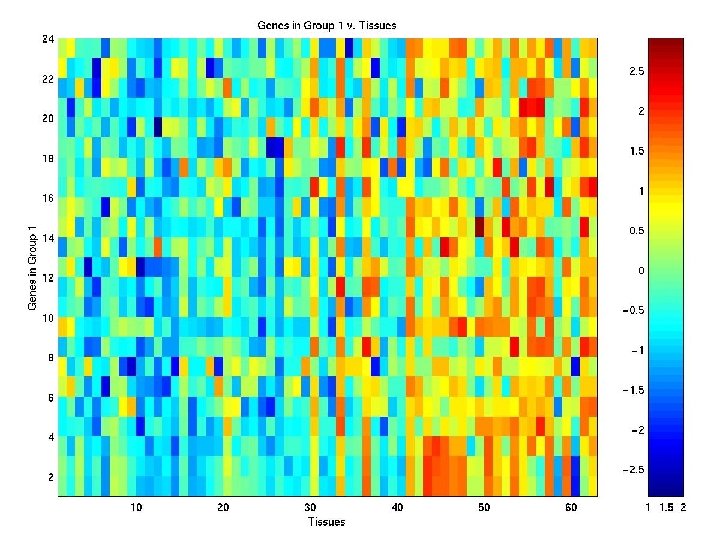

Figure 1: Error rates of the SVM rule with RFE procedure averaged over 50 random splits of colon tissue samples

Figure 2: Error rates of the SVM rule with RFE procedure averaged over 50 random splits of leukemia tissue samples

Figure 3: Error rates of Fisher’s rule with stepwise forward selection procedure using all the colon data

Figure 4: Error rates of Fisher’s rule with stepwise forward selection procedure using all the leukemia data

Figure 5: Error rates of the SVM rule averaged over 20 noninformative samples generated by random permutations of the class labels of the colon tumor tissues
 XIONG")
ADDITIONAL REFERENCES Selection bias ignored: XIONG et al. (2001, Molecular Genetics and Metabolism) XIONG et al. (2001, Genome Research) ZHANG et al. (2001, PNAS) Aware of selection bias: SPANG et al. (2001, Silico Biology) WEST et al. (2001, PNAS) NGUYEN and ROCKE (2002)
")
Error Rate Estimation Suppose there are two groups G 1 and G 2 c(y) is a classifier formed from the data set (y 1, y 2, y 3, ……………, yn) The apparent error is the proportion of the data set misallocated by c(y).

Cross-Validation From the original data set, remove y 1 to give the reduced set (y 2, y 3, ……………, yn) Then form the classifier c(1)(y ) from this reduced set. Use c(1)(y 1) to allocate y 1 to either G 1 or G 2.

Repeat this process for the second data point, y 2. So that this point is assigned to either G 1 or G 2 on the basis of the classifier c(2)(y 2). And so on up to yn.

Ten-Fold Cross Validation 1 Test 2 3 4 5 6 7 8 Training 9 10
. 632 estimator where B 1 is the bootstrap when")
BOOTSTRAP APPROACH Efron’s (1983, JASA). 632 estimator where B 1 is the bootstrap when rule the training sample. A Monte Carlo estimate of B 1 is where is applied to a point not in
 proposed the ERROR RATE ESTIMATOR where Mc. Lachlan (1977) proposed")
Toussaint & Sharpe (1975) proposed the ERROR RATE ESTIMATOR where Mc. Lachlan (1977) proposed w=wo where wo is chosen to minimize asymptotic bias of A(w) in the case of two homoscedastic normal groups. Value of w 0 was found to range between 0. 6 and 0. 7, depending on the values of
 where (relative overfitting rate) (estimate")
. 632+ estimate of Efron & Tibshirani (1997, JASA) where (relative overfitting rate) (estimate of no information error rate) If r = 0, w =. 632, and so B. 632+ = B. 632 r = 1, w = 1, and so B. 632+ = B 1

Ten-Fold Cross Validation 1 Test 2 3 4 5 6 7 8 Training 9 10

MARKER GENES FOR HARVARD DATA For a SVM based on 64 genes, and using 10 -fold CV, we noted the number of times a gene was selected. No. of genes 55 18 11 7 8 6 10 8 12 17 Times selected 1 2 3 4 5 6 7 8 9 10

MARKER GENES FOR HARVARD DATA No. of Times genes selected 55 1 18 2 11 3 7 4 8 5 6 6 10 7 8 8 12 9 17 10 tubulin, alpha, ubiquitous Cluster Incl N 90862 cyclin-dependent kinase inhibitor 2 C (p 18, inhibits CDK 4) DEK oncogene (DNA binding) Cluster Incl AF 035316 transducin-like enhancer of split 2, homolog of Drosophila E(sp 1) ADP-ribosyltransferase (NAD+; poly (ADP-ribose) polymerase) benzodiazapine receptor (peripheral) Cluster Incl D 21063 galactosidase, beta 1 high-mobility group (nonhistone chromosomal) protein 2 cold inducible RNA-binding protein Cluster Incl U 79287 BAF 53 tubulin, beta polypeptide thromboxane A 2 receptor H 1 histone family, member X Fc fragment of Ig. G, receptor, transporter, alpha sine oculis homeobox (Drosophila) homolog 3 transcriptional intermediary factor 1 gamma transcription elongation factor A (SII)-like 1 like mouse brain protein E 46 minichromosome maintenance deficient (mis 5, S. pombe) 6 transcription factor 12 (HTF 4, helix-loop-helix transcription factors 4) guanine nucleotide binding protein (G protein), gamma 3, linked dihydropyrimidinase-like 2 Cluster Incl AI 951946 transforming growth factor, beta receptor II (70 -80 k. D) protein kinase C-like 1

Breast cancer data set in van’t Veer et al. (van’t Veer et al. , 2002, Gene Expression Profiling Predicts Clinical Outcome Of Breast Cancer, Nature 415) These data were the result of microarray experiments on three patient groups with different classes of breast cancer tumours. The overall goal was to identify a set of genes that could distinguish between the different tumour groups based upon the gene expression information for these groups.

Breast tumours have a genetic signature. The expression pattern of a set of 70 genes can predict whether a tumour is going to prove lethal, despite treatment, or not. “This gene expression profile will outperform all currently used clinical parameters in predicting disease outcome. ” van ’t Veer et al. (2002), van de Vijver et al. (2002)

Number of Genes Error Rate for Top 70 Genes (without correction for Selection Bias as Top 70) Error Rate for Top 70 Genes (with correction for Selection Bias as Top 70) Error Rate for 5422 Genes (with correction for Selection Bias) 1 0. 50 0. 53 0. 56 2 0. 32 0. 41 0. 44 4 0. 26 0. 40 0. 41 8 0. 27 0. 32 0. 43 16 0. 28 0. 31 0. 35 32 0. 22 0. 35 0. 34 64 0. 20 0. 34 0. 35 70 0. 19 0. 33 - 128 - - 0. 39 256 - - 0. 33 512 - - 0. 34 1024 - - 0. 33 2048 - - 0. 37 4096 - - 0. 40 5422 - - 0. 44
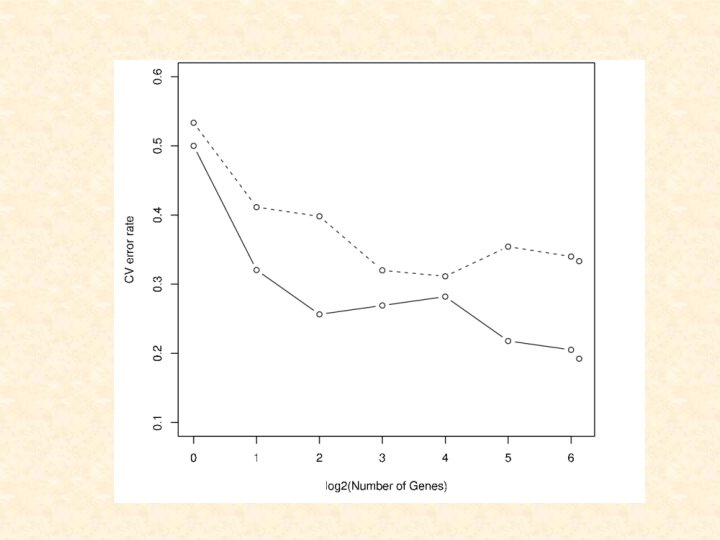
 considered a further 234 breast cancer tumours but")
van de Vijver et al. (2002) considered a further 234 breast cancer tumours but have only made available the data for the top 70 genes based on the previous study of van ‘t Veer et al. (2002)

Number of Genes From 70 genes From original 24481 genes (set missing values to 0) From original 24481 genes ( using KNN for missing values, k=10) 1 0. 29491525 0. 4023327 0. 4199797 2 0. 17288136 0. 3850913 0. 3825558 4 0. 20000000 0. 3747465 0. 3756592 8 0. 13220339 0. 3033469 0. 3061866 16 0. 10508475 0. 2314402 0. 2319473 32 0. 08474576 0. 2038540 0. 2240365 64 0. 09491525 0. 2038540 0. 1915822 70 0. 09491525 128 0. 1634888 0. 1600406 256 0. 1462475 0. 1507099 512 0. 1359026 0. 1438134 1024 0. 1324544 0. 1496957 2048 0. 1521298 0. 1364097 4096 0. 1481744 0. 1403651 8192 0. 1550710 0. 1605477 16384 0. 1683570 0. 1738337 24481 0. 1683570 0. 1772819

 The usual estimates of the class means")
Nearest-Shrunken Centroids (Tibshirani et al. , 2002) The usual estimates of the class means overall mean of the data, where and are shrunk toward the

The nearest-centroid rule is given by where yv is the vth element of the feature vector y and .

In the previous definition, we replace the sample mean the vth gene by its shrunken estimate v where of
 nearest-shrunken centroids and (ii) the SVM")
Comparison of Nearest-Shrunken Centroids with SVM Apply (i) nearest-shrunken centroids and (ii) the SVM with RFE to colon data set of Alon et al. (1999), with N = 2000 genes and M = 62 tissues (40 tumours, 22 normals)
 Overall Error Rates (b) Class-specific Error Rates")
Nearest-Shrunken Centroids applied to Alon data (a) Overall Error Rates (b) Class-specific Error Rates
 Overall Error Rates (b) Class-specific Error")
SVM with RFE applied to Alon data (a) Overall Error Rates (b) Class-specific Error Rates
- Slides: 55