Spinal Muscular Atrophy SMA Understanding Molecular aetiology treatment
 Understanding Molecular aetiology, treatment and Prenatal Diagnosis (PND) Mohammed AL-raqad")

















")



 for the treatment of Spinal Muscular Atrophy (SMA)")
 is an anti-sense oligonucleotide indicated for treatment of spinal muscular")
, which controls the mutations caused in the chromosome")


- Slides: 30
Spinal Muscular Atrophy (SMA) Understanding Molecular aetiology, treatment and Prenatal Diagnosis (PND) Mohammed AL-raqad
Summary of Key Issue For Topic • SMA phenotype shows a wide range from the very mild to the very severe. • SMA patients all show homozygous loss of SMN 1 gene • What factors are responsible for modulating the severity of the phenotype • Treatment for SMA.
• SMA is a severe neuromuscular disorder, characterised by degeneration of alpha motor neurons in the spinal cord which results in progressive symmetrical proximal muscle weakness • Inheritance autosomal recessive • Incidence 1 in 10 000 live births • Carrier rate 1 in 40 – 1 in 60
Clinical Classification • Type 1 - onset <6 months, can’t sit unaided death < 2 years • Type 2 - onset <18 months, can’t walk unaided death > 4 years • Type 3 - onset >18 months, walk but have proximal weakness, live till adulthood • Type IV / V- adult onset SMA, can be very mild
SMA Locus – 5 q 11. 2 • SMA types I, II & III all map to 5 q 11. 2 • This region is genetically complex containing multicopy repetitive sequences and pseudogenes (De novo rearrangements are identified in at least 2% SMA patients) • Characterisation of the smallest deletions in this region facilitated identification of the SMN 1 gene as the SMA determining gene
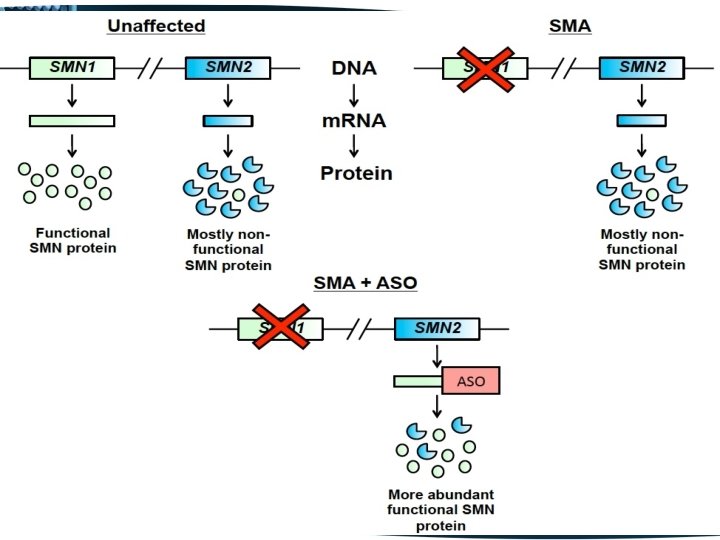
SMA is caused by homozygous loss of the SMN 1 gene • 95% patients are homozygous for the absence of SMN 1 (either whole gene, exon 7 or exons 7 & 8) • 5% are compound heterozygotes for the absence of SMN 1 exon 7 plus a more subtle intragenic mutation
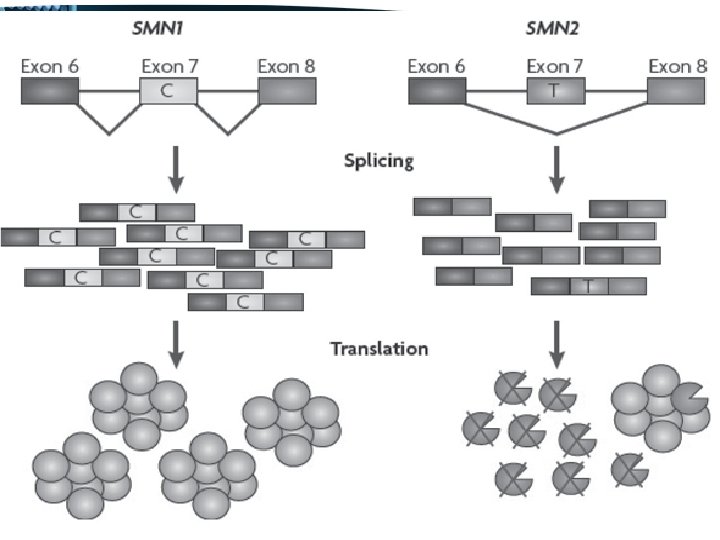
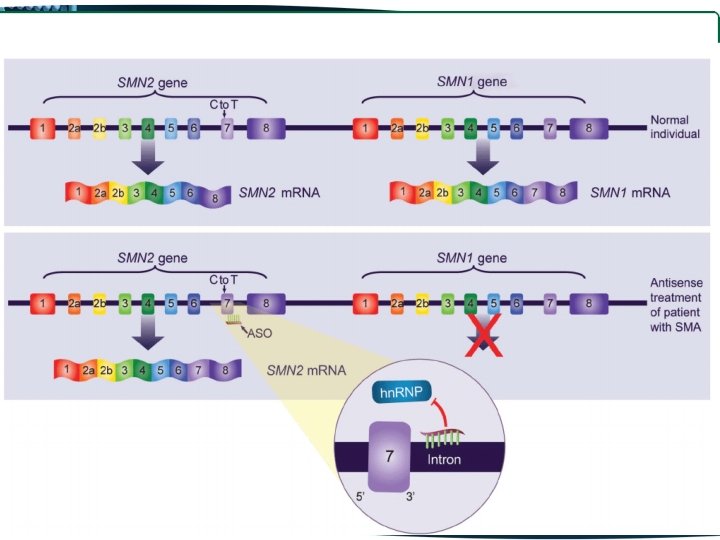
The SMN 1 gene is duplicated with a highly homologous copy called SMN 2 SMN 1 previously called SMN telomeric SMN 2 previously called SMN centromeric • Both SMN 1 and SMN 2 are transcribed • There are only 5 nucleotide differences between SMN 1 & SMN 2 • None of these differences alter amino acids
• One of these differences, a C to T change in SMN 2 exon 7 disrupts an exonic splice enhancer which results in the majority of SMN 2 transcripts lacking exon 7 SMN 1 produces full length transcript SMN 2 produces mainly truncated transcripts, lacking exon 7, producing a protein lacking the last 16 residues of the C terminal
Strong Evidence That SMN 2 Copy Number Modulates The Severity of the SMA Phenotype • The SMN 2 copy number varies in the normal population from 0 -3 copies (~ 10 -15% have 0 copies) • When SMN 1 is deleted / mutated, the SMN 2 gene cannot fully compensate for loss of expression of SMN 1. Milder patients with type II or type III SMA have been shown to have more copies of SMN 2 than type I patients. Harada et al 2002 found: SMN 2 copy no in type 1 patients = 2. 2 + 0. 6 SMN 2 copy no in type II & III patients = 3. 1 + 0. 3
Basic Genotype / Phenotype Hypothesis • Type I SMA is associated with SMN 1 gene deletion • Type II & III SMA have SMN 1 gene deletion plus gene conversion events that result in an increased number of SMN 2 copies, whereby SMN 2 gene is copied either partially or totally into the SMN 1 locus.
Prior et al 2002 describe 3 cases of asymptomatic individuals who have homozygous loss of SMN 1 • Carrier testing on father of type 3 SMA patient showed he had homozygous deletion of SMN 1 + 5 copies of SMN 2. He showed no symptoms & had a very active lifestyle • 3. 5 year old infant with type I SMA, had SMN 1 homozygous deletion + 2 copies SMN 2. Her younger brother was diagnosed prenatally, found to have SMN 1 homozygous deletion + 5 copies SMN 2. At 2 years, he is completely asymptomatic.
• Carrier testing of a 40 year old man revealed he had homozygous deletion of SMN 1 + 5 copies of SMN 2. His brother, who was diagnosed with type III SMA at aged 14, had exactly the same genotype • Prior et al 2002, speculate that: Expression levels consistent with 5 copies of the SMN 2 gene maybe enough to compensate for absence of the SMN 1 gene in some patients
Situation Is Further Complicated By the Finding That A Sub-group Of Type I SMA Patients Carry 3 Copies of SMN 2 • Harada et al 2002 investigated one such patient using RT-PCR. They found that the patient produced a large amount of truncated SMN 2 m. RNA species but only a trace of full length SMN 2 m. RNA, much less than expected from an individual with 3 copies SMN 2 • Authors speculate that this is due to alterations in the splicing mechanism which splices exon 7 from the transcipt
• This is backed up by other reports of haploid-identical siblings from SMA families with discordant phenotypes who show no difference in SMN 1 and SMN 2 gene copy numbers. • Conclude that though there is a general correlation of SMN 2 copy number with the severity of SMA, not all SMN 2 alleles are functionally equivalent • The genetic background influencing splicing mechanism of SMN 2 may be more critical in some patients and account for some intrafamilial variability.
Summary - SMA phenotype is modulated by various compensating factors • There is a general correlation of SMN 2 copy number with the severity of SMA - milder patients with type II / type III SMA typically have more copies of SMN 2 than type I patients. • 5 copies of the SMN 2 gene maybe enough to compensate for absence of the SMN 1 gene in some rare asymptomatic individuals • Not all SMN 2 alleles not functionally equivalent. Some SMA type I patients with 3 copies of SMN 2 actually produce very little full length SMN 2 transcript due to the genetic background influencing SMN 2 splicing mechanism
Summary - SMA phenotype is modulated by various compensating factors • Some intrafamilial variability in patients with an identical genotype may be due to differences in their genetic background influencing splicing mechanism of SMN 2 • Differences in the extent of SMN 1 deletion may also impact on the phenotype. The role of the NAIP gene which is deleted in <50% SMA type I patients is uncertain • Other modifying genes may play a role in modulating phenotype.
The SMN Protein • Ubiquitously expressed in cytoplasm & nucleus • SMN 1 self associates via its N & C terminus domains (SMN 2 protein lacking C terminus has reduced self association) • Mouse studies show that SMN 2 transcript is unstable in vivo
SMN May Play Role in Pre-m. RNA Splicing • SMN protein localised to nuclear structures called “gems” which are involved in the processing and metabolism of small nuclear ribonucleoproteins (Sn. RNPs). • Sn. RNPs are known to require activation from the inactive to the active form. SMN is thought to function in the reassembly and regeneration of these splicing components • Lack of functional SMN in patient motor neurons would impair their capacity to produce specific m. RNAs. These neurons would then become deficient in proteins necessary for normal growth, resulting in symptoms of the disease
Prenatal Diagnosis (PND)
06 07 08 09 10 11 12 13 14 15
06 07 08 09 10 11 12 13 14 15
6= 19% 26= 81%
Spinraza (nusinersen) for the treatment of Spinal Muscular Atrophy (SMA)
• Spinraza™ (nusinersen) is an anti-sense oligonucleotide indicated for treatment of spinal muscular atrophy (SMA) in paediatric and adult patients. • Developed by Ionis Pharmaceuticals, Spinraza was approved by the US Food and Drug Administration (FDA) under priority review in December 2016. • The European Medicines Agency (EMA) accepted the marketing authorisation application (MAA) for Spinraza in October 2016. • The Committee for Medicinal Products for Human Use (CHMP) of EMA has granted accelerated assessment designation to Spinraza.
Spinraza contains an anti-sense oligonucleotide (ASO), which controls the mutations caused in the chromosome 5 q. This selectively binds and targets RNA and regulates gene expression. It has the potential to enhance the amount of functional SMN protein in infants and children with SMA. The drug can be administered through intrathecal injection directly to the cerebrospinal fluid (CSF) around the spinal cord.
• A randomised, double-blind, sham-controlled clinical study conducted for 13 months on 121 patients with infantile-onset SMA. • The motor milestone response was measured by the Hammersmith Infant Neurological Examination (HINE). • Interim analysis results showed infants treated with Spinraza achieved the primary endpoint of motor milestone response compared with those who did not receive treatment. • Also, a smaller percentage of patients (23%) treated with Spinraza died compared with untreated patients (43%).
Thank you