Spektrometria mas MALDITOFTOF Kazimierz Dbrowski Katedra Chemii i

Spektrometria mas MALDI-TOF/TOF Kazimierz Dąbrowski Katedra Chemii i Technologii Polimerów Wydział Chemiczny PW Warszawa, 16. 04. 2012

Najważniejsze odkrycia w historii spektrometrii mas Rok 1911 1918 1930 1942 1946 Odkrycie Zbudowanie pierwszego spektrometru mas Opracowanie źródła jonów EI Zastosowanie spektrometrii mas w chemii organicznej Pierwszy sprzedany spektrometr mas Analizator czasu przelotu (TOF) Nazwiska badaczy Joseph John Thomson Arthur Jeffrey Dempster R. Conrad Consolidated Energy Corporation William E. Stephens 1953 Spektrometr o podwójnym ogniskowaniu E. G. Johnson i A. O. Nier 1953 1958 Kwadrupolowy analizator masy Połączenie spektrometru mas z chromatografem gazowym (GC) W. Paul i H. Steinwedel 1966 Sekwencjonowanie peptydów przy pomocy spektrometru mas 1966 1968 1981 1983 1984 K. Biemann, C. Cone, B. R. Webster i B. P. Arsenault Jonizacja Chemiczna (CI) B. Munson i F. H. Field Jonizacja przez Elektrorozpylanie (ESI) Malcom Dole Metoda jonizacji przez bombardowanie szybkimi atomami (FAB) Michael Barber Opracowanie metody jonizacji przez desorpcję laserem przy udziale Koichi Tanaka, Michael Karas i Franz matrycy – MALDI (Nagroda Nobla z Chemii w 2002 roku) Hillenkamp G. C. Statford, P. E. Kelly, J. E. P. Pułapka jonowa Syka, W. E. Reynolds i J. F. J. Todd Wykorzystanie elektrorozpylania (ESI) do analizy biopolimerów Gall Lydia (ZSRR), John Fenn (USA) (Nagroda Nobla w 2002 roku)

Podstawowe definicje Spektrometr masowy – instrument pozwalający na precyzyjny pomiar stosunku masy do ładunku (m/z) analizowanych substancji. Rozdzielczość spektrometru – wartość liczbowa informująca o możliwości rozróżnienia na widmie masowym pików o zbliżonych masach. W przypadku pojedynczego piku wartość określająca dokładność oznaczenia masy cząsteczkowej (atomowej) substancji analizowanej. Jeśli spektrometr masowy w danym momencie analizy posiada rozdzielczość R=1000 istnieje możliwość rozróżnienia pików o m/z=1000 oraz m/z=1001. Dla izolowanego piku rozdzielczość definiuje jego szerokość połówkową, tzn. dla R=1000 i piku o m/z=1000 stosunek jego wysokości do szerokości w 0, 5 wysokości wynosi co najmniej 10 (H/L 0, 5 h>=10) Jon molekularny – jon obdarzony ładunkiem (ładunkami) powstający w wyniku fragmentacji próbki w źródle jonów Jon fragmentacyjny – jon powstały w wyniku spontanicznej fragmentacji substancji (np. podczas jonizacji metodą EI) lub uzyskany techniką tandemowej spektrometrii masowej. Dostarcza informacji o strukturze substancji analizowanej. Addukt - jon powstały poprzez przyłączenie do analizowanej substancji np. jonu sodowego Proteom - PROTEin complement of the gen. OME (ogół białek kodowanych przez genom) Dalton - jednostka masy, dokładnie odpowiada 1, 0000 na skali mas atomowych Dekonwolucja - uzyskanie rzeczywistej masy substancji z widma pików wielokrotnie zjonizowanych Matryca - niskocząsteczkowe związki organiczne absorbujące promieniowanie lasera.
 - jonizacja elektronami MALDI (Matrix-Assisted Laser Desorption/Ionization) -")
Najczęściej stosowane skróty EI (Electron Impact) - jonizacja elektronami MALDI (Matrix-Assisted Laser Desorption/Ionization) - jonizacja laserem wspomagana matrycą ESI (Electrospray Ionization) - jonizacja przez rozpylanie w polu elektrycznym HPLC (High Performance Liquid Chromatography) - wysokosprawna chromatografia cieczowa) MS/MS (Tandem Mass Spectrometry) - tandemowa spektrometria masowa TOF (Time of Flight Analyser) - analizator czasu przelotu PSD (Post Source Decay) - rozpad poza źródłem jonów m/z - stosunek wartości masy do liczby ładunków DIOS (Desorption/Ionization on Porous Silicon) - desorpcja/jonizacja na porowatym krzemie ICP (Inductively Coupled Plasma) - jonizacja plazmą wzbudzoną indukcyjnie

Idea działania spektrometru mas • źródło jonów – urządzenie, w którym następuje jonizacja cząsteczek przy użyciu różnorodnych technik, z których część prowadzi do pękania wiązań chemicznych na skutek czego dochodzi do ich podziału na mniejsze fragmenty. Inne techniki powodują tylko naładowanie cząsteczek bez ich fragmentacji, • analizator – w którym wcześniej powstałe jony ulegają rozdziałowi na podstawie stosunku ich masy do ładunku. • detektor – urządzenie "zliczające" jony napływające z analizatora.
![Jonizacja próbki Twarda technika jonizacji Jon molekularny typu [M+. ] Bogate widmo fragmentacyjne Łagodna](http://slidetodoc.com/presentation_image_h/d7e0562882c3f2c7a8fe028e6d810d77/image-6.jpg "Jonizacja próbki Twarda technika jonizacji Jon molekularny typu [M+. ] Bogate widmo fragmentacyjne Łagodna")
Jonizacja próbki Twarda technika jonizacji Jon molekularny typu [M+. ] Bogate widmo fragmentacyjne Łagodna technika jonizacji Pozorny jon molekularny [M+H]+ lub [M-H]Brak fragmentacji

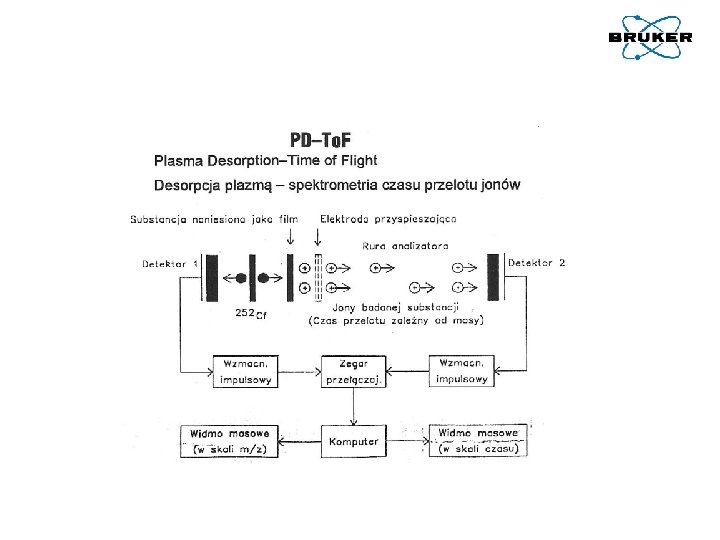
Jonizacja próbki cd.

Jonizacja próbki – lasery używane w technice MALDI Nitrogen laser: pro: well structured energy profile contra: slow (maximum 50 Hz) Nd: YAG laser: pro: fast (up to 1000 Hz) contra: Gaussian energy profile (non-structured) Smartbeam/Smartbeam II (modified Nd: YAG laser): pro: fast (up to 1000 Hz) pro: well structured energy profile A. Holle, A. Haase, M. Kayser, J. Höhndorf, Journal of Mass Spectrometry, 41, 705 -716 (2006)

Matryce Procesy zachodzące pod wpływem impulsu laserowego Absorpcja promieniowania głównie przez materiał matrycy. Odparowanie próbki na głębokość 2 -3 l i wyrzucenie strumienia gazów prostopadle do jej powierzchni. Dysocjacja termiczna matrycy. Tworzenie jonów (głównie H+, Na+, K+). Reakcje jonów z badaną substancją i matrycą. Możliwe drogi: - dysocjacja termiczna z utworzeniem pary kation-anion - oderwanie elektronu - oderwanie bądź przyłączenie protonu - przyłączenie kationu bądź anionu

Matryce Pożądanymi cechami matrycy MALDI są: • dość niska masa cząsteczkowa, co sprzyja łatwemu odparowaniu, ale wystarczająco duża, by odparowanie nastąpiło przed pomiarem, np. w czasie przygotowywania próbki; • rozpuszczalność w rozpuszczalniku kompatybilnym z analitem; • kwasowość, by ułatwić protonowanie cząsteczek analitu; • obecność grup polarnych (hydrofilowych) w cząsteczce, co umożliwia rozpuszczanie matrycy w roztworach wodnych; • stabilność w warunkach wysokiej próżni; • wspomaganie jonizacji analitu; • zdolność intensywnej absorpcji promieniowania UV lasera; zwykle wymóg ten spełnia związek, posiadający układ sprzężonych wiązań podwójnych C=C (dlatego często matrycami są pochodne aromatycznych kwasów karboksylowych, często nienasyconych, np. kwasu cynamonowego).

Matryce Sinapic acid Kwas 3 -5 -dimetoksy-4 -hydroksy cynamonowy SINA proteiny Dithranol 1, 8 -Dihydroksyantracen-9(10 H)-on DIT polimery Gentisic acid Kwas 2 -5 -dihroksy benzoesowy DHB peptydy α-Cyano-4 -hydroxycinnamic acid Kwas α-Cyano-4 -hroksy cynamonowy CHCA, α-CHCA Peptydy, lipidy 2 -(4 -Hydroxyphenylazo)benzoic acid Kwas 2 -(4 -hydroksyfenylazo)- benzoesowy HABA Peptydy, polimery 2, 4, 6 -Trihydroxyacetophenone 2, 4, 6 -trihroksy acetofenon THAP oligonukleotydy

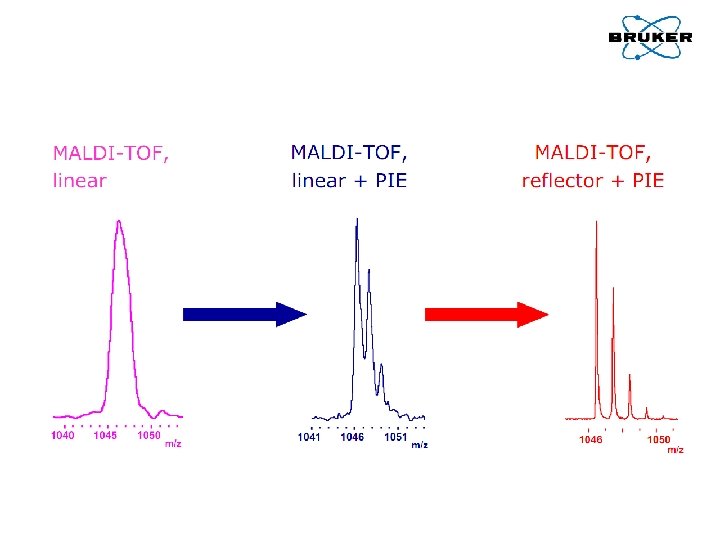
Tryb liniowy

Tryb liniowy Jak zwiększyć rozdzielczość? • Pulsed ion extraction – polepszenie ogniskowania jonów • Optymalizacja przygotowania próbki – homogenizacja • Użycie reflektronu

Optymalne przygotowanie próbki
 to: 1. 2. Metoda wysychającej kropli")
Najczęściej stosowane metody nanoszenia próbki (analitu i matrycy) to: 1. 2. Metoda wysychającej kropli (dried droplet method) – jednowarstwowa: Przyrządza się osobno roztwór próbki i roztwór matrycy w tym samym rozpuszczalniku, lub – jeśli to niemożliwe – w dwóch kompatybilnych; niekiedy jeszcze używa się trzeciego roztworu - środka kationizującego, np. soli metalu (możliwe są różne rozpuszczalniki i stężenie). Wszystkie roztwory miesza się, a uzyskaną mieszaninę (0, 5÷ 1 μl) umieszcza na płytce MALDI (MALDI target) i pozostawia do wyschnięcia na powietrzu. Wadą tej metody jest powolne wysychanie próbki, co może prowadzić do rozdzielenia kryształów matrycy próbki i soli kationizującej. Modyfikacjami tej metody są: zastosowanie odparowywania próżniowego lub odparowywanie w strumieniu ultraczystego azotu. W obu przypadkach otrzymuje się drobniejsze kryształy, lepszą rozdzielczość i powtarzalność oraz intensywność sygnałów. Metoda cienkiej warstwy (thin-layer method) – dwuwarstwowa: Roztwór matrycy w odpowiednim rozpuszczalniku (np. dla CHCA – roztwór nasycony w acetonie) nanosi się na płytkę MALDI i pozwala mu się wyschnąć, otrzymując cienką warstwę matrycy. Następnie 1 μl roztworu analitu nanosi się na wierzch uzyskanej powierzchni matrycy i suszy. 3. Nanoszenie przez rozpylanie. Wariantami tej metody są: osadzanie strumieniem powietrza (air spray deposition) i osadzanie przez elektrosprej (electrospray sample depositon). 4. Metoda mieszania ciał stałych (solid/solid sample preparation): Metoda polega na bardzo dokładnym mieszaniu drobno sproszkowanych matrycy i próbki (bez rozpuszczalnika) i prasowaniu całości w pastylkę. Stosuje się ją dla niektórych poliamidów, nierozpuszczalnych w pospolitych rozpuszczalnikach organicznych

Epot = ze. U Ekin = 1/2 mv 2 ze. U = 1/2 mv 2 v = (2 ze. U/m)1/2 t = L×(1/2 e. U)1/2×(m/z)1/2

Tryb reflektronu

Tryb reflektronu

Profil izotopowy

Profil izotopowy Masa monoizotopowa Masa średnia C 41 H 69 N 13 O 14 S [M+H]+: 1000. 4880 [M+H]+: 1001. 1409 C 112 H 164 N 29 O 34 S 2 [M+H]+: 2524. 1510 [M+H]+: 2525. 8196 C 253 H 363 N 55 O 75 S [M+H]+: 5404. 6075 [M+H]+: 5407. 9984

Kalibracja tof= tdelay + tacc + tdrift F = E q = M a d = a/2 tacc 2 E = U/d q = z e tacc = d √(2 m/Uze) tdrift= L √(m/2 ze. U) tof=tdelay + d √(2 m/Uze) + L √(m/2 ze. U) tof=tdelay + (d √(2/Ue)+L √(1/2 Ue)) × √(m/z) t = C 0+C 1√(m/z)
 Ion path in")
MALDI–To. F/To. F Ion path in TOF 1 region (linear TOF) Ion path in TOF 2 region (reflector. TOF) Ion source 1 = MALDI ion source Ion source 2 = LIFT re-acceleration cell PCIS = Timed ion gate PLMS = Post LIFT meta stable suppressor

MALDI–To. F/To. F

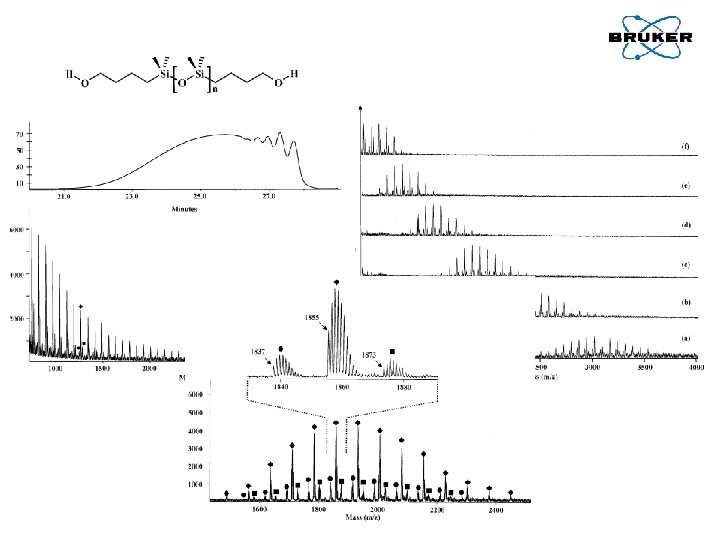
MALDI–To. F w badaniach polimerów Za pomocą MALDI można wyznaczyć następujące parametry polimerów: • • • Mn, Mw, PD grupy końcowe polimeru budowę kopolimeru Montaudo G, J. Polym. Sci. A: Polym. Chem. 34, 1996, 439 -447

C B A A 9 B 2 C A 8 B 2 C A 9 BC A 8 B 3 C A 10 B 2 C A 10 BC A 9 B 3 C A 11 BC A 7 B 3 C A 6 B 4 C A 8 B 4 C A 7 B 4 C A 10 C A 11 C
 Polisacharydy, teflon,")
Polimery polarne: poliwęglany, politlenki olefin Polikwasy, poliestry, polimery słabo polarne (np. polistyren) Polisacharydy, teflon, poliolefiny mass discrimination effect
 fractionated with acetone, hexanes, methylene chloride, THF, and")
MALDI mass spectra of a poly(alkylthiophene) fractionated with acetone, hexanes, methylene chloride, THF, and chloroform Liu, J. ; Loewe, R. S. ; Mc. Cullough, R. D. Macromolecules 1999, 32, 5777 -5785.

Wskazówki dla PT Klientów • • • Badane mogą być związki stałe, ewentualnie ciecze nielotne (ciśnienie w komorze pomiarowej < 10 -7 Torr). Do pomiaru używamy zazwyczaj 5 mg substancji. Próbki można dostarczać albo już odważone (proszę podać masę próbki), lub w większych pojemnikach. W razie tzw. wyższej konieczności do pomiaru wystarczy: substancji typu biologicznego (np. peptydu) 0, 1 - 10 pmol, polimeru 50 - 200 pmol. Można również dostarczyć próbkę w postaci roztworu w dowolnym, ale koniecznie lotnym i niekorodującym rozpuszczalniku (prosimy o podanie stężenia roztworu !) Możliwy jest pomiar próbki nierozpuszczalnej, ale wówczas musi być ona w postaci możliwie drobnego proszku. Należy jednak podkreślić, że wyniki takiego pomiaru będą znacznie gorszej jakości, niż próbek rozpuszczalnych. Kategorycznie odmawiamy przyjmowania podejrzanych, dymiących cieczy zawierających stężone lotne kwasy (np. HCI, HNO 3) oraz substancji korodujących typu POCI 3, wolne aminy, fenole, merkaptany itd. Aby proces tworzenia jonów przebiegał możliwie wydajnie, próbka powinna być jak najczystsza. W szczególności nie powinna zawierać: • nadmiaru soli nieorganicznych z grupy litowców i miedziowców, • detergentów !!!, • buforów, zwłaszcza fosforanowych, • substancji silnie alkalizujących (reagują z matrycą), W przypadku polimerów, co do których zachodzi podejrzenie, że mogą zawierać frakcje znacznie różniące się masą cząsteczkową ( np. Mn = 1 500 i Mn = 30 000) niezbędne jest wstępne rozdzielenie próbki na frakcje. Prosimy o podanie spodziewanej masy cząsteczkowej, bądź przynajmniej zakwalifikowanie próbki do jednego z następujących przedziałów: < 5000; 5000 -20000; > 20000 Do badanej próbki (lub grupy próbek) prosimy dołączyć „Zlecenie wykonania badania MALDI-To. F” (druk dostępny na stronie: www. ch. pw. edu. pl/~maldi )

Polecana literatura • M. Karas, D. Bachmann, F. Hillenkamp; Analytical Chemistry, 57, 2935 -2939 (1985) • K. Tanaka, H. Waiki, Y. Ido, S. Akita, Y. Yoshida, T. Yoshida; Rapid Communications in Mass Spectrometry, 2, 151 -153 (1988) • R. C. Beavis, B. Chait, K. G. Standing; Rapid Communications in Mass Spectrometry, 3, 233 -237 (1989) • M. Karas, M. Glückmann, J. Schäfer; Journal of Mass Spectrometry, 35, 1 -12, (2000) • R. Zenobi and R. Knochenmuss; Mass Spectrometry Reviews, 17, 337 -366 (1998) • G. Montaudo, M. S. Montaudo, and F. Samperi, “ Mass Spectrometry of Polymers ”, ed. G. Montaudo and R. P. Lattimer, 2002 , CRC Press, Boca Raton, FL, 419.

Dziękuję za uwagę…
- Slides: 34