SINDROME HEMOFAGOCTICO ANDRS MANN UPCP HOSPITAL PUERTO MONTT




 • Reingreso UPCP (13/9) para tratamiento con pulsos de")
 •")

 • Hemorragia digestiva masiva • Fallece el 8/11/2007")

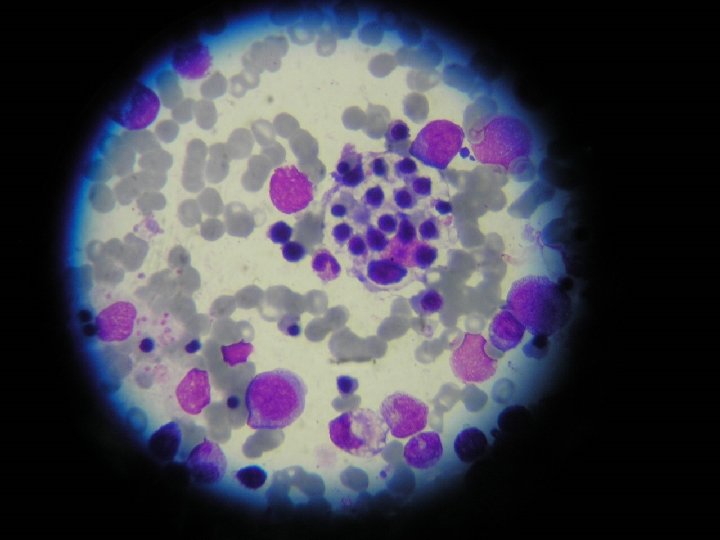
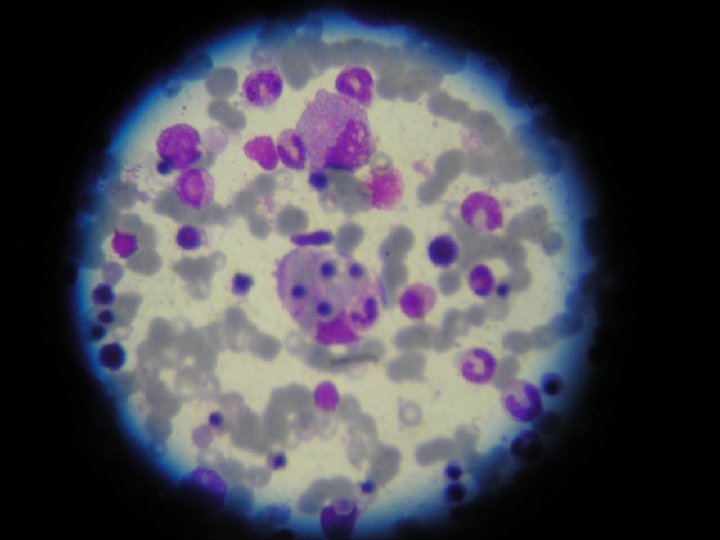
 Citología medular característica de Síndrome hemofagocítico. (26/19) Hiperplasia")
 Normal (14/9) Normal (28/9) Miocardiopatía dilatada Insuficiencia")



 • Familiar (FHLH o HLH primaria): Autosómica recesiva Asociada a disminución")
 • Sindrome hemofagocítico secundario (HLH secundario o s. HLH) Sindrome de")

 • • Clínico y de laboratorio (5 de 8): Fiebre Esplenomegalia")
 Diagnóstico molecular Mutaciones en el gen de la perforina (10 q")
 Terapia de sostén Antibióticos de")
; 150 mg/m")
 • Etopósido c/2 sem • Dexametasona pulsos 10 mg/m")











- Slides: 37
SINDROME HEMOFAGOCÍTICO ANDRÉS MANÉN UPCP HOSPITAL PUERTO MONTT JULIO 2008
CASO CLÍNICO • • F. A. V. C. SEXO FEM FECHA DE NAC: 8/7/2005 EDAD AL INGRESO: 2 años 1 mes DOMICILIO: Cochamó INGRESO: 30/8/2007 EGRESO: 8/11/2007
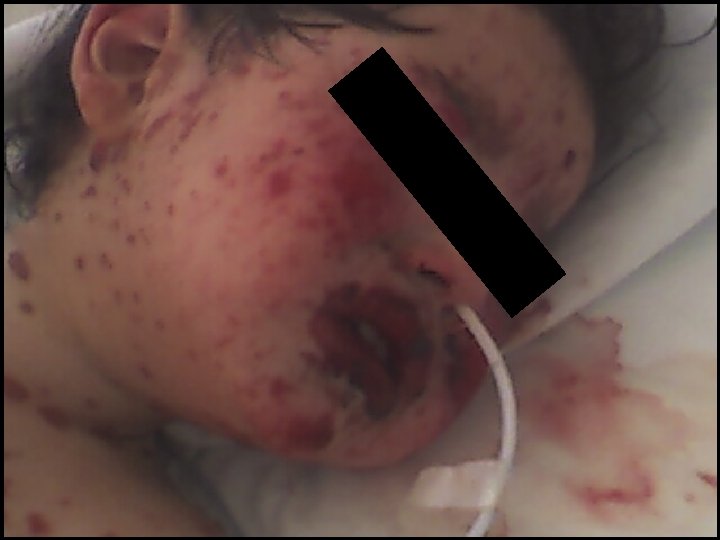
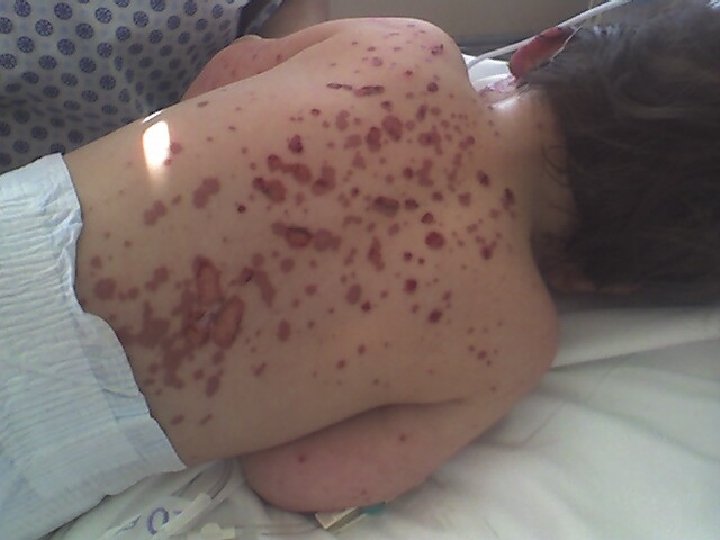
CASO CLÍNICO • Sin antecedentes mórbidos ni familiares relevantes • Fiebre de 5 días previo al ingreso • Pancitopenia y elevación de Transaminasas • Erupción máculo papular confluente en cara y EE
CASO CLÍNICO • Diagnóstico: Eritema Multiforme Mayor • ECO Cardio Normal • Ingreso UPCP (5/9): Tratamiento con Gammaglobulina iv • Egreso UPCP (7/9) en tratamiento con Cefuroximo por OMA • Mejoría hematológica
CASO CLÍNICO • Mielograma (11/9) • Reingreso UPCP (13/9) para tratamiento con pulsos de Metilprednisolona • Mielograma compatible con Sindrome Linfohistiocitosis Hemofagocítico • Fibrinógeno disminuido, TG aumentados • IGM para Epstein Barr positivo
CASO CLÍNICO • Tratamiento con Ganciclovir • Tratamiento con Dexametasona y Ciclosporina (21/9) • Insuficiencia Cardiaca por Miocardiopatía dilatada y Aneurisma coronario derecho (28/9). • Segundo tratamiento con Gammaglobulina iv. • Soporte ventilatorio y vasoactivo (Milrinona y Dopamina)
CASO CLÍNICO • Mejoría clínica y de laboratorio a las 3 semanas de tratamiento • Recaída desde el 24/10 • TAC de tórax, TAC y ECO de abdomen (25/10) normales • Mielograma (26/10) Hemofagocitosis
CASO CLÍNICO • Inicio Etopósido (2/11) • Hemorragia digestiva masiva • Fallece el 8/11/2007
EXÁMENES 30/8 5/9 13/9 21/9 28/9 12/10 23/10 29/10 2/11 6/11 8/11 Hto. 29 26, 9 22, 3 27, 3 22, 5 23, 6 21, 5 20, 4 25 20, 2 22, 7 Hb 9, 8 9, 3 7, 7 8, 6 7, 3 8, 3 7, 4 7 7, 7 7 8 Leuco 2800 6520 1890 700 1000 14800 6100 2660 1600 1460 599 Plaq 84000 145000 22600 17000 8000 104000 133000 74100 25000 12400 12600 GOT 263 157 460 341 40 33 95 183 526 TG 502 434 373 93 161 141 99 Fbg 91 170 205 Ferriti na >1000 669 >1000 >30000
OTROS EXÁMENES • • Mielograma: (11/9) Citología medular característica de Síndrome hemofagocítico. (26/19) Hiperplasia de serie eritropoyética Hipoplasia serie granulopoyética Hemofagocitosis HAV Ac, HBs Ag, HCV Ac, negativos (5/9) Ig. M virus Epstein Barr, positivo (5/9) Ag/Ac VIH, negativo (13/9)
OTROS EXÁMENES • • ECO Cardio: (4/9) Normal (14/9) Normal (28/9) Miocardiopatía dilatada Insuficiencia Tricuspídea leve Aneurisma coronaria derecha (3/10) Aneurisma coronaria derecha en regresión Diskinesia de zona septal de VI TAC de Tórax Normal, TAC y ECO de Abdomen moderada hépatoesplenomegalia (25/10)
SINDROME HEMOFAGOCÍTICO
Nomenclatura • Histiocitosis: grupo de alteraciones que tienen en común la proliferación de células del sistema fagocítico mononuclear.
Clasificación • Alteraciones de comportamiento biológico variable: Alteraciones relacionadas con Células Dendríticas (Histiocitosis de células de Langerhans) Alteraciones de los Macrófagos • Alteraciones malignas
Linfohistiocitosis Hemofagocítica (HLH) • Familiar (FHLH o HLH primaria): Autosómica recesiva Asociada a disminución en el gatillo de la apoptosis 20 a 40 % por mutación en el gen de la perforina (defecto en citotoxicidad en NK y linfocitos T) Inicio y recaídas gatilladas por infecciones Incidencia: 1, 2/1. 000 niños/año (1: 50. 000 NV)
Linfohistiocitosis Hemofagocítica (HLH) • Sindrome hemofagocítico secundario (HLH secundario o s. HLH) Sindrome de activación marofágica (sistema fagocítico mononuclear) Por infecciones (VAHS o IAHS) Por enfermedades reumatológicas Por enfermedades malignas (MAHS) Por alteraciones metabólicas (sindrome de sobrecarga lipídica)
Cuadro clínico • • Fiebre Hepatoesplenomegalia Citopenia Hipertrigliceridemia Hipofibrinogenemia Transaminasas y Ferritina elevadas Linfadenopatía, ictericia, erupción cutánea y edema
Criterios diagnósticos A) • • Clínico y de laboratorio (5 de 8): Fiebre Esplenomegalia Citopenia (2 o más series): Hb < 9; Plaquetas < 100. 000; Neutrófilos < 1. 000 Hipertrigliceridemia ( 265 mg/dl) y/o hipofibrinogenemia ( 150 mg/dl) Hemofagocitosis en MO, bazo o ganglios Ferritina 500 mcg/l Receptor de IL-2 soluble 2. 400 U/ml Actividad de células NK baja o ausente
Criterios diagnósticos B) Diagnóstico molecular Mutaciones en el gen de la perforina (10 q 21) Mutaciones en h. Munc 13 -4 (17 q 25)
Tratamiento • • • Manejo Agudo (semanas 1 -8) Terapia de sostén Antibióticos de amplio espectro Profilaxis con CTM (5 mg/Kg de TMT) 2 -3 veces por semana Profilaxis antimicótica vo Tratamiento antiviral Ig iv (0, 5 g/Kg) c/4 semanas
Tratamiento • Etopósido: 150 mg/m 2 iv 2 veces/sem (sem 1 -2); 150 mg/m 2 iv 1 vez/sem (sem 3 -8) • Dexametasona: 10 mg/m 2/día (sem 1 -2); 5 mg/m 2/día (sem 3 -4); 2, 5 mg/m 2/día (sem 5 -6); 1, 25 mg/m 2/día (sem 7); suspender en sem 8 • Ciclosporina A: 6 mg/Kg/día ajustar según nivel plasmático
Tratamiento de Mantención (sem 940) • Etopósido c/2 sem • Dexametasona pulsos 10 mg/m 2 por 3 días c/2 sem • Ciclosporina A para mantener niveles plasmáticos de 200 mcg/L
Tratamiento Trasplante de MO: Es el único tratamiento curativo en el FHL
Estados clínicos • • • Respuesta clínica: Ausencia de fiebre Reducción de esplenomegalia Plaquetas > 100. 000 Fibrinógeno normal Descenso de niveles de Ferritina (25 %)
Estados clínicos • • • Resolución: Ausencia de fiebre Ausencia de esplenomegalia Sin citopenia Triglicéridos normales Ferritina <500 mcg/L LCR normal
Estados clínicos Enfermedad activa Reactivación
Literatura GONZALEZ M. , Benito, ROA A. , Johanna y SCHMIDT S. , Nadia. Síndrome de activación macrofagico en pediatría: A propósito de cuatro casos. Rev. chil. pediatr. , abr. 2005, vol. 76, no. 2, p. 183 -192. ISSN 0370 -4106. VERDUGO L. , Patricia, RODRIGUEZ Z. , Natalie, TORDECILLA C. , Juan et al. Síndrome hemofagocítico secundario en pediatría: Experiencia clínica en ocho casos. Rev. chil. pediatr. , ago. 2005, vol. 76, no. 4, p. 397 -403. ISSN 0370 -4106.
Literatura Nahum, Elhanan MD; Ben-Ari, Josef MD; Stain, Jeremiya MD; Schonfeld, Tommy MD Hemophagocytic lymphohistiocytic syndrome: Unrecognized cause of multiple organ failure. Pediatric Critical Care Medicine. 1(1): 51 -54, July 2000. Steinberg, Orna MD *; Yacobovich, Joanne MD +; Dgany, Orly Ph. D +; Kodman, Yona MSc +; Livni, Gilat MD *; Rachmel, Avinoam MD *; Stein, Jerry MD +; Yaniv, Isaac MD +; Tamary, Hannah MD + Prolonged Course of Familial Hemophagocytic Lymphohistiocytosis. Journal of Pediatric Hematology/Oncology. 28(12): 831 -833, December 2006.
Literatura Morrell, Dean S. MD; Pepping, Marie A. MD; Scott, J. Paul MD; Esterly, Nancy B. MD; Drolet, Beth A. MD Cutaneous Manifestations of Hemophagocytic Lymphohistiocytosis. Archives of Dermatology. 138(9): 1208 -1212, September 2002.
Literatura Gonzalez-Llano, Oscar; Jaime-Perez, Jose; Cantu-Rodriguez, Olga; Mancias-Guerra, Consuelo; Gutierrez-Aguirre, Homero; Herrera-Garza, Jose; Gomez-Almaguer, David Successful father-to-son stem cell transplantation in a child with hemophagocytic lymphohistiocytosis using a reducedintensity conditioning regimen. European Journal of Haematology. 77(4): 341 -344, October 2006.
FIN