SINDROME DE WOLFRAM 1 INTRODUCCIN El Sndrome de










- Slides: 12
SINDROME DE WOLFRAM
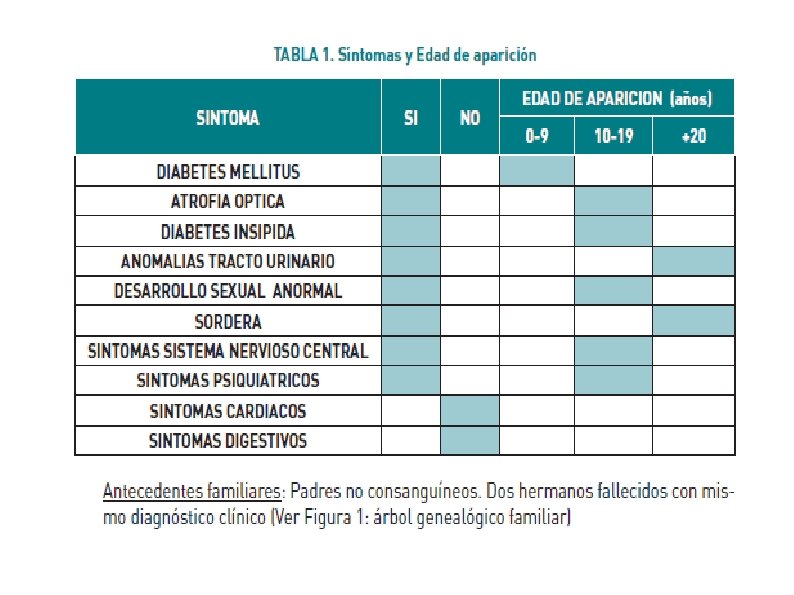
1. INTRODUCCIÓN: El Síndrome de Wolfram es una enfermedad neurodegenerativa rara ( prevalencia 1/160000 ). DIDMOAD ( Diabetes insípida, Diabetes melitus tipo I, Atrofia óptica y sordera ). También alteraciones del aparato urinario, atrofia gonadal, síntomas neurológicos y alteraciones psiquiatricas.
Se han identificado dos genes responsables del WFS, con un patrón de herencia autosómico recesivo: - El gen WFS 1, que codifica para la Wolframina proteína localizada en el retículo endoplasmático, que interviene en la homeostásis cálcica. ( Es el más frecuente ) - El gen CISD 2, que codifica para la 2 CDGSH proteína intermembrana del retículo endoplasmático.
2. EXPOSICIÓN DEL CASO 2. 1 Anamnesis y exploración física Mujer de 48 años de edad remitida desde la consulta de neurología con diagnóstico clínico sugestivo de WFS. Antecedentes personales: Diabetes Mellitus Tipo I de difícil control, ceguera por atrofia óptica, diabetes insípida, sordera neurosensorial bilateral, vejiga neurógena, antecedentes de crisis tónico-clónicas en tratamiento. Síndrome de ansiedad, distimia, en tratamiento psiquiátrico de evolución crónica.
2. 2 Informe del Laboratorio: INFORME BIOQUÍMICO (Sección de Bioquímica General y Sección de Orinas). Sin hallazgos relevantes. 2. 3 Diagnóstico diferencial:
2. 4 Exploraciones complementarias: Estudio genético molecular del WFS (gen WFS 1, OMIM 606210) obteniéndose como resultado, dos mutaciones en heterocigosis: - En el exón 4 del gen WFS 1, que produce un codón de parada ( V 142 fs. X 251 ) -En el exón 8 del gen WFS 1, que produce un codón de parada en el AA 520 ( Q 520 X ) Ambas mutaciones son responsables de la síntesis de una proteína anómala o inexistente. El hallazgo de las dos mutaciones WFS 1 confirma el diagnóstico clínico de sospecha de WFS 1.
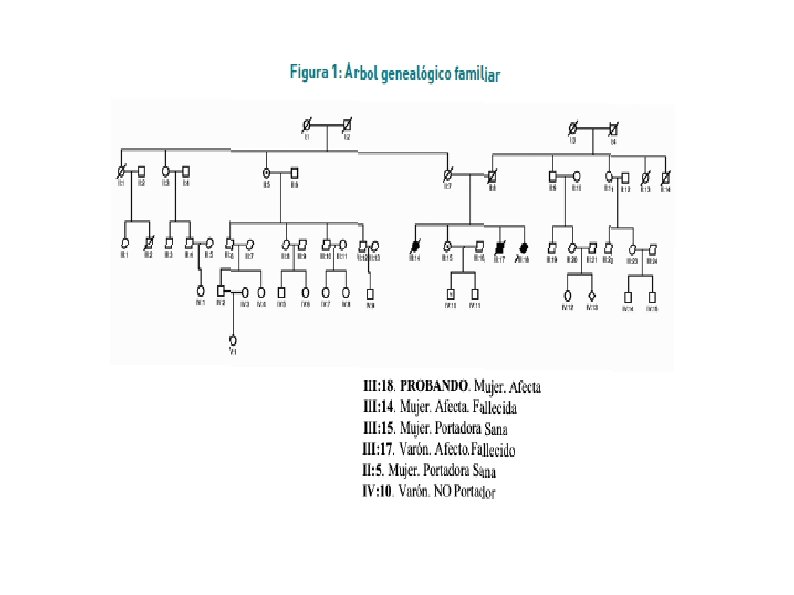
2. 5 Diagnostico definitivo La presencia en la paciente de DIDMOAD es compatible con el diagnóstico clínico de WFS el cual se ha confirmado al identificarse dos mutaciones patogénicas en heterocigosis: c. 409_424 dup 16 (V 142 fs. X) / c. 1558 C>T (Q 520 X) en el exón 4 y en el exón 8 respectivamente del gen WFS 1. La hermana de la paciente resultó ser portadora de la mutación Q 520 X. La tía por rama paterna resultó ser portadora de la otra mutación (V 142 fs. X), quedando identificada la segregación familiar de ambas mutaciones.
2. 6 Evolución La paciente es totalmente dependiente para las actividades cotidianas de la vida diaria debido a su condición de invidente y alteraciones neurológicas y recibe un seguimiento clínico y bioquímico muy frecuente.
3. Discusión El debút del cuadro clínico es una Diabetes Mellitus insulino dependiente no autoinmune en edad juvenil. Ante un cuadro clínico así deberá hacerse un buen diagnostico diferencial y no olvidarse del WFS ya que muchas veces los síntomas que acompañan a la diabetes se interpretan como complicaciones de ésta y no como entidades nosológicas por sí mismas. El diagnóstico precoz y adecuado tratamiento ( paliativo ) mejorará la vida del paciente. Consejo genético para la familia.
GRACIAS