SICKLE CELL ANEMIA Sicklecell disease SCD also known


, also known as sickle-cell anaemia (SCA) and drepanocytosis is a hereditary")














 and activated Partial Thromboplastin Time (a. PTT) a. PTT")
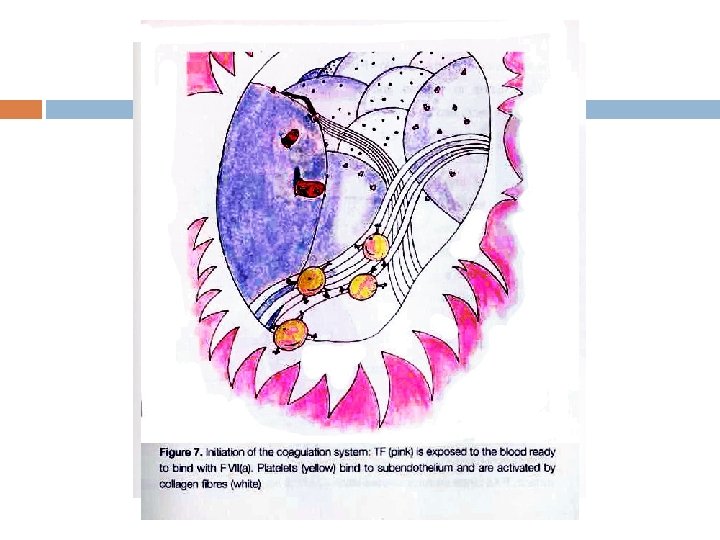

 usually forms around atherosclerotic plaques.")





























- Slides: 56
SICKLE CELL ANEMIA
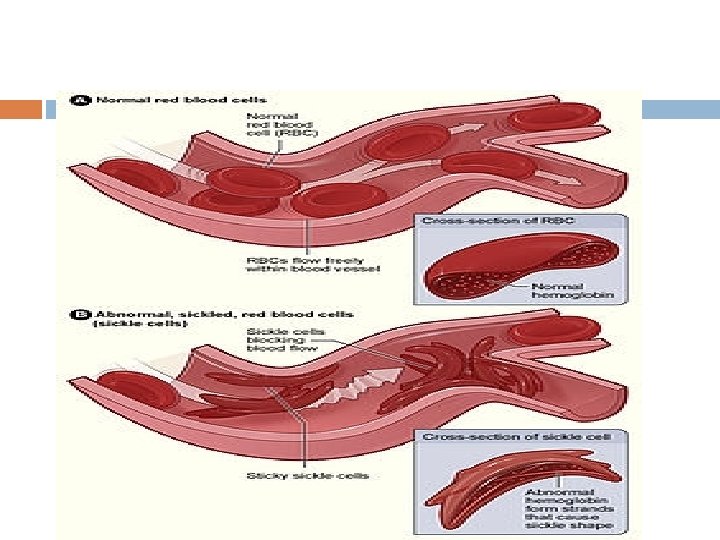
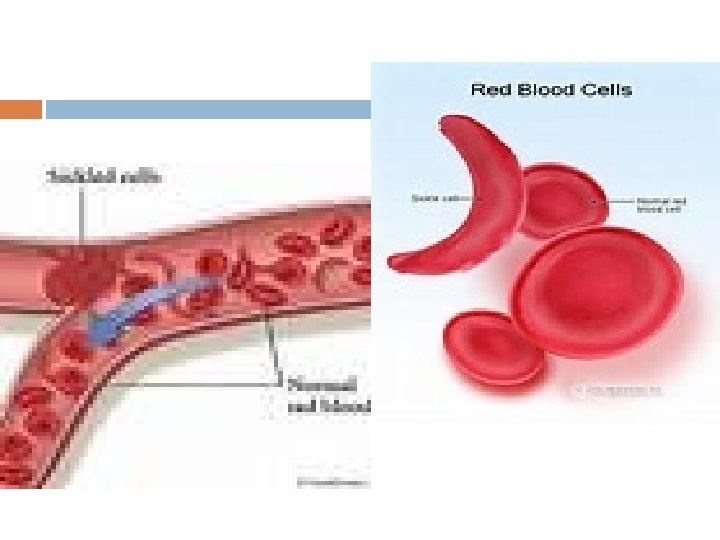
Sickle-cell disease (SCD), also known as sickle-cell anaemia (SCA) and drepanocytosis is a hereditary blood disorder, characterized by an abnormality in the oxygen-carrying haemoglobin molecule in red blood cells
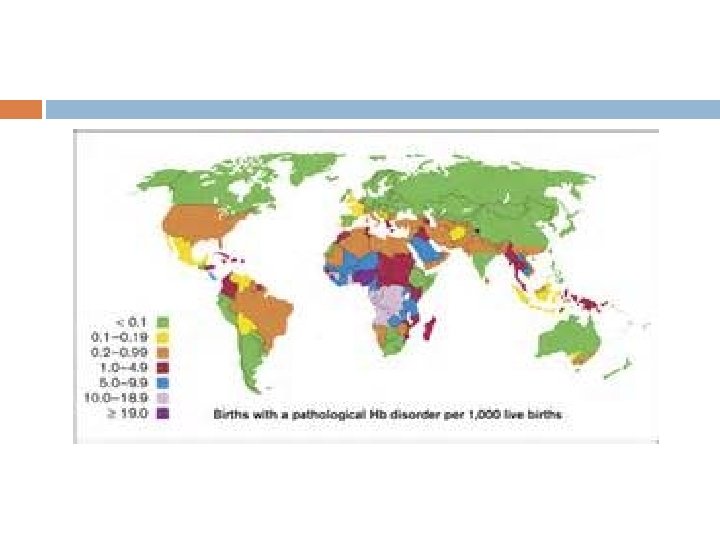
Almost 300, 000 children are born with a form of sickle-cell disease every year mostly in sub-Saharan Africa, but also in other countries such as the West Indies and in people of African origin elsewhere in the world.
Epidemiology The highest frequency of sickle cell disease is found in tropical regions, particularly sub. Saharan Africa, tribal regions of India and the Middle-East. Africa: Three quarters of sickle-cell cases occur in Africa around 2% of newborns in Nigeria were affected by sickle cell anaemia.
150, 000 affected children born every year in Nigeria alone. The carrier frequency ranges between 10% and 40% across equatorial Africa. United States: The prevalence of the disease in the United States is approximately 1 in 5, 000, mostly affecting Americans of Sub-Saharan African descent,
In the United States, about 1 out of 500 African. American children and 1 in every 36, 000 Hispanic-American children born will have sickle-cell anaemia. Sickle cell trait occurs among about 1: 12 African. Americans and 1: 100 Hispanic-Americans. It is estimated that 2. 5 million Americans are heterozygous carriers for the sickle-cell trait
Middle East: In Saudi Arabia about 4. 2% of the population carry the sickle-cell trait and 0. 26% have sickle -cell disease. The highest prevalence is in the Eastern province where approximately 17% of the population carry the gene and 1. 2% have sickle-cell disease.
India: Sickle-cell disease is common in the tribal people of central India who share a genetic linkage with the african race, where the prevalence has ranged from 9. 4 to 22. 2% in endemic areas of madhya pradesh, rajasthan and chhattisgarh.
In 2013 it resulted in 176, 000 deaths up from 113, 000 deaths in 1990. The condition was first described in the medical literature by the American physician James B. Herrick in 1910, and in the 1940 s and 1950 s contributions by Nobel prize-winner Linus Pauling made it the first disease where the exact genetic and molecular defect was elucidated.
Prognosis About 90% of patients survive to age 20, and close to 50% survive beyond the fifth decade. In 2001, according to one study performed in Jamaica, the estimated mean survival for sickle-cell patients was 53 years old for men and 58 yearsold for women with homozygous SCD.
Vaso-occlusive crisis The vaso-occlusive crisis is caused by sickleshaped red blood cells that obstruct capillaries and restrict blood flow to an organ resulting in ischaemia, pain, necrosis and often organ damage
Painful crises are treated with hydration, analgesics, and blood trsnsfusion. pain management requires opioid administration at regular intervals until the crisis has settled.
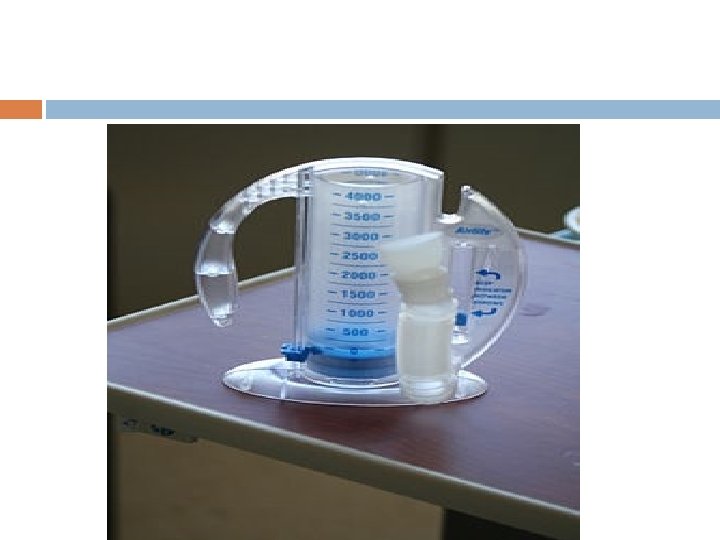
For more severe crises, most patients require inpatient management for intravenous opioids; patient-controlled analgesia devices. Incentive spirometry, a technique to encourage deep breathing to minimise the development of atelectasis, is recommended.
CNS Crisis in SCD
Bleeding Clotting Hemostasis
Lab Tests: Prothrombin time (PT) and activated Partial Thromboplastin Time (a. PTT) a. PTT is initiated with phospholipid (PL), calcium and silica. nl time to clot = 31 -55 sec. PT is initiated by adding thromboplastin (PL + TF) and calcium to plasma. nl time to clot = 10 -16 sec.
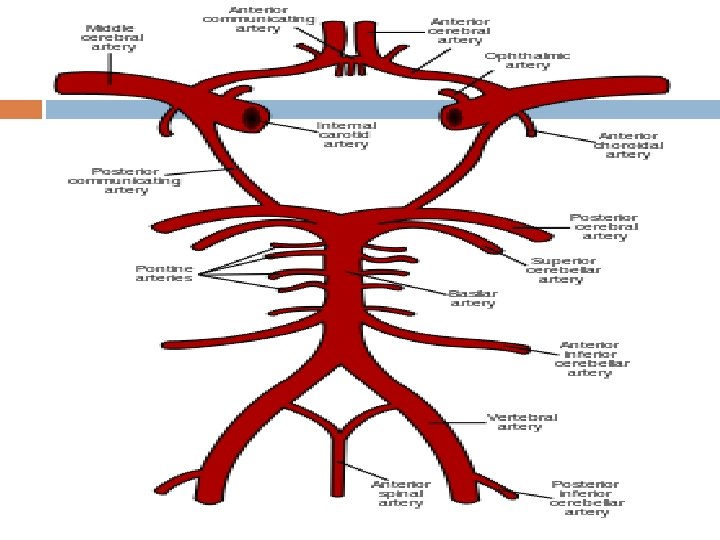
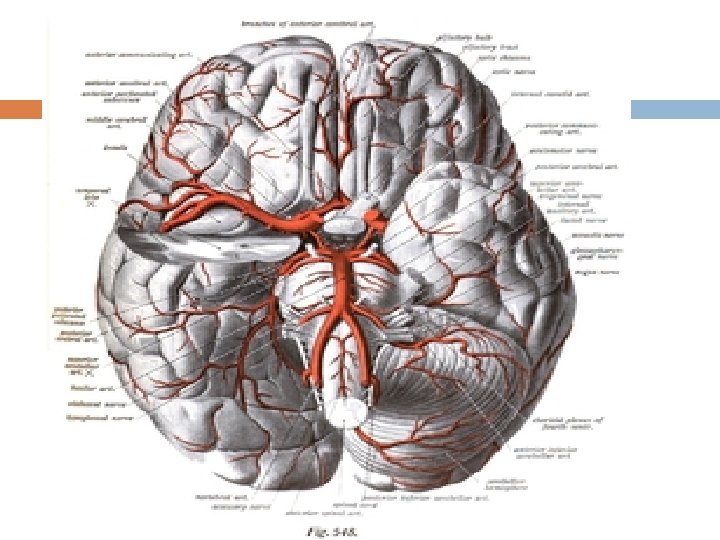
Thrombotic stroke In thrombotic stroke a thrombus (blood clot) usually forms around atherosclerotic plaques. Two types of thrombosis can cause stroke: • Large vessel disease involves the common and internal carotids, vertebral, and the Circle of Willis.
noninflammatory vasculopathy, Moyamoya disease and fibromuscular dysplasia. Small vessel disease involves the smaller arteries inside the brain: branches of the circle of Willis, middle cerebral artery, stem, and arteries arising from the distal vertebral and basilar artery.
Moyamoya syndrome is a disease in which certain arteries in the brain are constricted. Blood flow is blocked by the constriction, and also by blood clots (thrombosis). A collateral circulation develops around the blocked vessels to compensate for the blockage, but the collateral vessels are small, weak, and prone to hemorrhage, aneurysm and thrombosis. On conventional X-ray angiography, these collateral vessels have the appearance of a "puff of smoke(moya)" in Japanese). [
Sickle cell anemiawhich can cause blood cells to clump up and block blood vessels, can also lead to stroke. A stroke is the second leading under 20 who suffer from killer of people sickle-cell
Hemorrhagic Intracranial hemorrhage intracerebral hemorrhage
Prevention and Management of Stroke The major breakthrough in the field of sicklerelated brain injury has been the unprecedented success of transcranial Doppler ultrasonography (TCD) to identify asymptomatic patients at high risk of stroke, coupled with chronic transfusion therapy to prevent it.
The evidence for TCD screening and preventive treatment is strong and compelling, but there are still important unanswered questions regarding the implications of “silent infarcts” found in the magnetic resonance images (MRIs) of asymptomatic individuals,
acute stroke occurs at a rate of ~600/100, 000 patient-years. resulting in a prevalence approaching 10% by the age of 50. It is no surprise that acute stroke became an early target for clinical intervention and investigation.
that acute mortality and morbidity could be minimized with the aggressive use of exchange transfusion at presentation. that patients with one stroke are at very high risk of recurrence;
that chronic maintenance transfusion could prevent recurrence.
TCD is a reproducible, noninvasive technique to find narrowed internal carotid or middle cerebral arteries in asymptomatic children by detecting a signature high-flow pattern. Children with elevated velocities (≥ 200 cm/sec) have an astonishingly high rate of stroke: ~10, 000/100, 000 patient-years
When asymptomatic children with abnormally high flow velocities demonstrated by using screening TCD are preemptively treated with maintenance transfusion, 90% of strokes are prevented.
the following is considered to be rational practice: 1 -Screen all children with SS ages 2– 16 using TCD. 2 -Repeat annually for children with normal studies, and every 6 months for those in the “conditional” range.
Once confirmed with a repeat study, children with abnormal TCD should be placed on a chronic maintenance transfusion program using blood matched for ABO, C, D, E, and Kell antigens, and timed and dosed to keep the [Hb. S] ≤ 30%.
Two other important alternatives are being studied, and hold considerable promise: 1 - hydroxyurea (HU), 2 - bone marrow transplant (BMT).
Silent stroke causes no immediate symptoms, but is associated with damage to the brain. Silent stroke is probably five times as common as symptomatic stroke. About 10– 15% of children with SCD suffer strokes, with silent strokes predominating in the younger patients
Bone marrow transplants have proven to be effective in children. Bone marrow transplants are the only known cure for SCD. However, bone marrow transplants are difficult toobtain because of the specific HLA typing necessary. Ideally, a twin family member (syngeneic) or close relative (allogeneic) would donate the bone marrow necessary for transplantation.
Gene therapy In 2001 it was reported that sickle-cell disease had been successfully treated in mice using gene therapy. The researchers demonstrated this method of gene therapy to be a more permanent means to increase the production of therapeutic Hb. F. Phase 1 clinical trials of gene therapy for sickle cell disease in humanswere started in 2014.
Transfusion Therapy in Sickle Cell Disease transfusion can be life-saving and prevent progressive organ damage. INDICATIONS FOR TRANSFUSIONS: to correct the low oxygen-carrying capacity caused by severe anemia, and to improve microvascular perfusion by decreasing the proportion of sickle red cells in the circulation
-Management of severe anemia: Hemoglobin values of less than 5 gm/dl or a 20 percent fall below the base line during an acute illness are common transfusion triggers,
Acute splenic sequestration transfusions are indicated when the hemoglobin drops by more than 2 gm/dl from the steady state.
Transient red cell aplasia: red cell transfusions are indicated for those who become symptomatic or whose hemoglobin value falls 2 gm/dl below baseline.
Acute chest syndrome many patients can be treated with a simple red cell transfusion. In severe cases, exchange transfusion/red cell pheresis is recommended.
Stroke: Chronic transfusion therapy early exchange transfusions may improve perfusion and oxygenation to brain tissue, thus limiting damage.
PREPARATION FOR GENERAL ANESTHESIA A multi-institution study recently compared perioperative complications among patients with sickle cell disease undergoing major surgery (e. g. cholecystectomy). Patients were randomized to an aggressive transfusion arm (decrease hemoglobin S to below 30 percent) or to a conservative transfusion arm (hemoglobin S approximately 60 percent; hemoglobin correctedto 10 gm/dl).
The recommendation from this study is that all sickle cell disease patients undergoing major surgery be prepared in advance with transfusion to correct their anemia to a hemoglobin of approximately 10 gm/dl and hemoglobin S percent to approximately 60 percent. No standard practice guidelines have been developed for patients undergoing minor procedures or for patients with hemoglobin SC disease.
four individuals to have won more than one Nobel Prize Marie Curie, Frederick Sanger Linus Carl Pauling John Bardeen