Sequence Alignment techniques Definition A sequence alignment is


































, GDE, Clustal, GCG/MSF, RSF. •")


- Slides: 41
Sequence Alignment techniques
Definition • A sequence alignment is a way of arranging the sequences of DNA, RNA, or protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationship between the sequences.
Sequence Alignments ? 1. I have just sequenced something. What is known about the thing I sequenced? 2. I have a unique sequence. Is there similarity to another gene that has a known function? 3. I found a new protein in a lower organism. Is it similar to a protein from another species? 4. I have decided to work on a new gene. The people in the field will not give me the plasmid. I need the complete c. DNA sequence to perform RT-PCR. 5. I wish to perform molecular modeling of the proteins sequence which has significant similarity to sequence of a protein for which the 3 D structure is available.
Sequence alignments • Pair wise • Multiple
Pairwise protein sequence alignment • definition: compare pairs of sequences and search for series of characters that are in the same order • sequences in rows with identical (or similar) characters in same columns and non-identical (non-similar) characters either in same column (mismatch) or opposite a gap HCKLTFGKWFTSEW | ||| | KCGPTFGRIACGEM ----TFGK-----||| ----TFGR------ Global - entire sequences aligned up to both ends Local - most similar sub-regions of sequences aligned (islands of similarity)
Methods of pairwise sequence alignment… • dot matrix - all possible matches between sequence residues are found; used to compare two sequences to look for regions where they may align; very useful for finding indels and repeats in sequences; can be used as a first pass to see if there is any similarity between sequences • dynamic programming - mathematically guaranteed to find optimal alignment (global or local) between pairs of sequences; very computationally expensive - # of steps increases exponentially with sequence length
Dot matrix method 1 - one sequence listed along top of page and second sequence listed along the side 2 - move across row and put dot in any column where the character is the same 3 - continue for each row until all possible character matches between the sequences are represented by dots 4 - diagonal rows of dots reveal sequence similarity (can also find repeats and inverted repeats off the main diagonal) H C G E T F G R W F T P E W K C G P T F G R I A C G E M • • • • 5 - isolated dots represent random similarity unrelated to the alignment •
Protein sequence Alignments… • Dot matrix method not a convenient method • Manual alignment of sequences ? • For sequence of length N, about 22 N/√ 2 N alignments are possible (for n=300, 10179 alignments!) • Mathematical solution: Dynamic programming (nothing to do with computer!)
Protein sequence Alignments… • In naturally occurring conserved proteins certain amino acids are favorably replaced in the process of natural selection. • Based on these observations and mutations substitution matrices have been generated. • For example: – BLOSUM (Block Substitution Matrices) matrices: BLOSUM 40, BLOSUM 60 etc. – PAM (Point Accepted Mutation) matrices: PAM 80, PAM 120, PAM 250 • These matrices are used by various protein sequence alignment algorithms.
Dynamic programming • a dot matrix shows regions of similarity but not path that connects disjointed regions i. e. the optimal alignment which is the ultimate goal of pairwise sequence comparison • dynamic programming was applied to sequence alignment by Needleman & Wunsch to achieve this end • dynamic programming is a general class of optimization solutions that finds best solutions by breaking down large intractable problems into smaller pieces and then solving • ultimately a sequence or ‘path’ of subproblem scores that yields the highest overall score is chosen as the optimal solution for the entire problem
Dynamic programming & sequence alignment • overall problem is broken down into subproblems of aligning each residue of one sequence to each residue of the other • choose the best solution to the problem among the three options of (1) - aligning residues (2) - introducing gap in sequence 1 or (3) - introducing gap in sequence 2 • each high scoring choice rules out two low scoring choices - this is critical in reducing the overall space of alignments needed to be evaluated (essence of time saving) • the algorithm use a matrix similar to the dot matrix with sequences on the top and left axes • at each position in the matrix the algorithm computes the best score and stores a pointer from the previous position from where the highest score was derived • finally a ‘trace back’ step is performed where the highest scoring path along the pointers is traced - this represents the optimal alignment
Dynamic programming & sequence alignment: Steps… • Two sequences are arranged in a matrix table. • Initial GAP penalties (d) are listed in the first row or column. • First values of substitutions scores (Si, j) are filled in the table using substitution matrices • The simple matrix table is converted to dynamic programming table using the following mathematical equation. • Hi, j = max { (Hi-1, j-1 + Si, j), (Hi-1, j – d), (Hi, j-1 –d) }
GAP H G S A Q V -8 -16 -24 -32 -40 -48 GAP 0 K -8 -1 -9 -16 -24 -31 -39 T -16 -9 -3 -7 -15 -23 -30 E -24 -16 -11 -3 -8 -13 -21 0 -3 0 -1 2 -3 A -32 -24 -15 -10 2 -6 -13 E -40 -32 M -48 -39 -23 H -3 K -31 -15 G 0 T -23 S-6 -1 E -14 A 42 A -4 -4 Q -3 E 5 1 -2 -2 -1 -1 -2 -2 0 -2 -2 1 0 2 1 -1 0 5 1 -1 0 • Hi, j = max { (Hi-1, j-1 + Si, j), (Hi-1, j – d), (Hi, j-1 –d) } -2 1 0 V M
sequence 1 sequence 2 score M - N M G S 6 -12 1 M M 6 N G 0 A L D R 0 -3 A - L S D R 1 -12 -3 S T 1 D R T E 0 -1 T T 3 = = -5 -5
Which matrix to use? • PAM 120 for general use • PAM 60 for close relations • PAM 250 for distant relations • BLOSUM 62 for general use • BLOSUM 80 for close relations • BLOSUM 45 for distant relations • When comparing closely related proteins one should use lower PAM or higher BLOSUM matrices, for distantly related proteins higher PAM or lower BLOSUM matrices.
• Global alignment algorithms : Needleman and Wunsch • Local Alignment algorithms: Smith. Waterman local alignment http: //www. ebi. ac. uk/Tools/emboss/align/
Needleman S. B. and Wunsch C. D. 1970. J. Mol. Biol. 48: 443 -453 Smith T. F. and Waterman M. S. 1981. J. Mol. Biol. 147: 195 -197 Eddy, S. R. 2004. Nature Biotechnology 22: 909 - 910
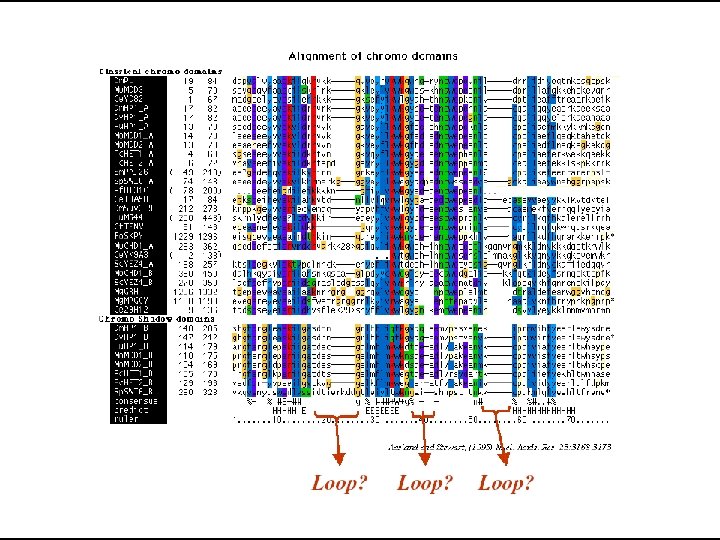
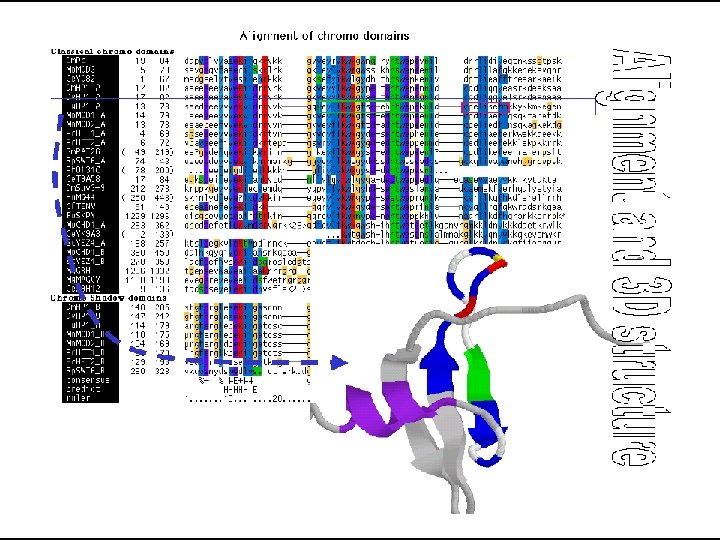
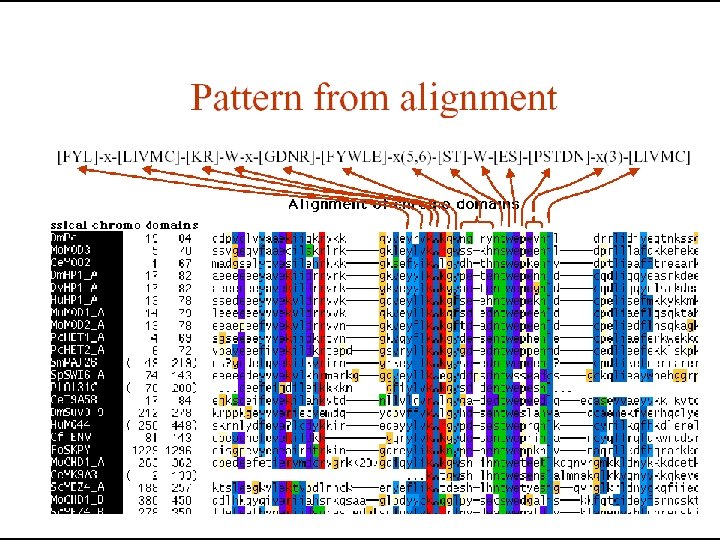
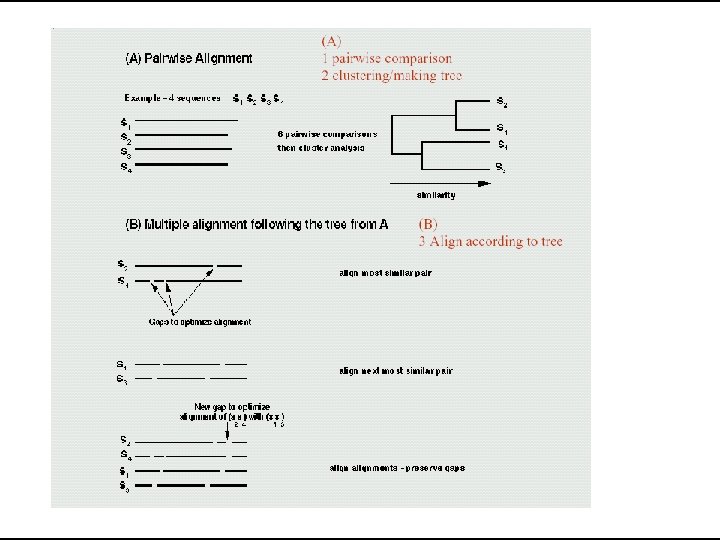
Multiple Sequence Alignment
Multiple Sequence Alignment
Clustal • Most widely used algorithm for MSA • Available in different forms Clustal. W, Clustal. X • Different Output formats • Apart from standalone it is also available in: • BIOEDIT, GCG, EMBOSS, Macvector etc.
Clustal. W • Formats: NBRF/PIR, EMBL/Swiss. Prot, Pearson (Fasta), GDE, Clustal, GCG/MSF, RSF. • Output formats: same as above + Phylip
Web server: http: //www. ebi. ac. uk/clustalw/index. html Align few sequences by default parameters. Change parameters like GAP penalties and note the changes in alignment outputs. Exercise: Make a dynamic programming matrix for a protein sequence of length 7. Use BLOSUM 40 matrix to generate a dynamic programming matrix using the mathematical equation given in the presentation. Trace back the path of maximum scores and obtain optical alignment(s)
Exercise to be submitted by Thursday • Go to http: //expasy. org/tools/randseq. html • Generate a random protein sequence of length 25 amino acids of average amino acid composition • Draw a dot plot. Identify regions of similarities, repeats, inverted repeats. • Submit the record to me.