SEMINARIO DE INDUCCION ASIGNATURA HEMATOLOGIA DIEGO FERNANDO LOPEZ




























 es normal, pero la concentración hemoglobínica corpuscular")


 es la anomalía")
















 Tipo Clínica Hemograma Leucemia linfoide aguda Fatiga –Fiebre Sangrado, infecc. Adenopatías Dolores –SNC")
















 se producen de forma descontrolada unas células linfoides")


- Slides: 71
SEMINARIO DE INDUCCION ASIGNATURA HEMATOLOGIA DIEGO FERNANDO LOPEZ MUÑOZ DOCENTE UNIVERSIDAD CATOLICA DE MANIZALES FACULTAD DE SALUD PROGRAMA BACTERIOLOGIA
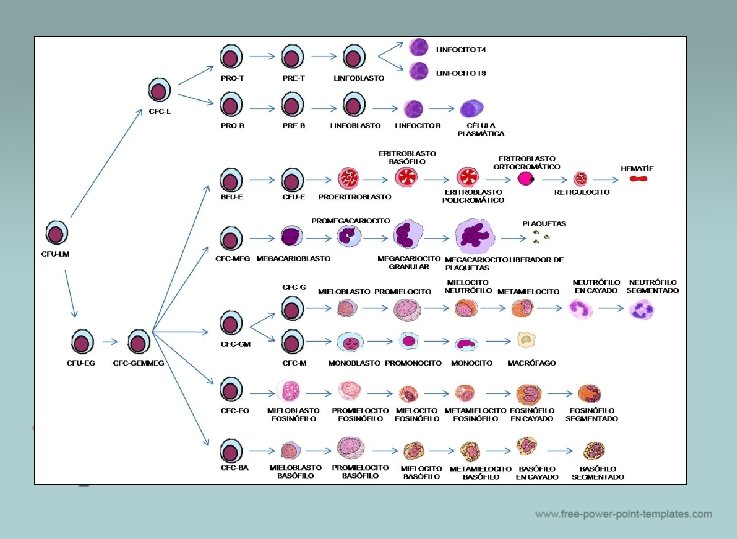
HEMATOPOYESIS
PROCESO Dinámico Proliferación Diferenciación Maduración Células sanguíneas Inicia en la stem cell hematopoyética, la cual asegura la producción permanente, adecuada y funcional de los elementos formes de la sangre
LAS INTERLEUCINAS. Son proteínas solubles de bajo peso molecular mediadoras de crecimiento celular, inflamación, inmunidad, diferenciación y reparación, entre otras actividades.
Microambiente Hematopoyético Consiste en una estructura tridimensional, altamente organizada, de células del estroma y sus productos (matriz extracelular, citosinas, quimocinas) que regulan la localización y fisiología de las células hematopoyéticas.
v MIELOPOYESIS: v Proceso de generación, desarrollo y maduración de los componentes mieloides de la sangre. Estos componentes son aquellos cuyo proceso de maduración, se inicia y completa en la médula ósea. ü LINFOPOYESIS: ü La linfopoyesis es el proceso del desarrollo hematopoyético, en el que se forman los Linfocitos y células Natural Killers (NK), a partir de una célula madre hematopoyética(Hematopoyetica Stem Cell). Cada una de las células que se forman (Linfocitos B, Linfocitos T y Cél. Natural Killers), tiene una génesis y proceso de maduración independiente, que culmina en distintos órganos. Linfocitos B lo hacen en el bazo, Linfocitos T lo hacen en el timo y los Linfocitos NK lo hacen de un progenitor hematopoyético pero deben recibir el estimulo de la IL 15
ERITROPOYESIS Ø Proceso por el cual se producen las células rojas de la sangre. Se estimulada por la disminución de O 2 en circulación, que es detectado por los riñones, los que secretan la hormona eritropoyetina. Esta hormona estimula la proliferación y diferenciación de precursores de células rojas Otros reguladores IL 3: crecimiento de células progenitoras hematopoyéticas Andrógenos: estimulan la eritropoyesis a través de un incremento en la producción renal de eritropoyetina Estrógenos atraviesan la membrana celular para llegar al núcleo, en el que se encargan de activar o desactivar determinados genes, regulando la síntesis de proteínas Insulina: Los glucocorticoides estimulan la eritropoyesis, dando lugar a un aumento en el número de hematíes. Pero también inhiben la inflamación como el cortisol Hormonas tiroides: Efecto hematopoyético: aumenta la eritropoyetina
LA SANGRE ü Tejido conectivo porque se origina de células similares. ü Representa cerca del 8% del peso corporal total del hombre adulto, y tiene un volumen de cinco a seis litros en un hombre tamaño promedio.
v Circula por los vasos sanguíneos. v El corazón es el órgano encargado de que pueda llegar a todos los tejidos del cuerpo. v Transporta nutrientes y O 2 a las células de manera directa o indirecta. v Transporta CO 2 y otros desechos de las células. v Distribuye hormonas y sustancias reguladoras. v Ayuda al mantenimiento de la homeostasis actuando como buffer, participa en la coagulación y es termorreguladora. v Transporta células y agentes humorales del sistema inmune.
GENERALIDADES DE LAS PATOLOGÍAS HEMATOPOYETICAS ü Anemias ü Leucemias ü Síndromes mielodisplasicos ü Síndromes mieloproliferativos ü Gamapatias monoclonales ü Linfomas
Anemia se define como la disminución de la masa de hemoglobina circulante. Se origina por dos causas principales. v. Presencia de un numero insuficientes de eritrocitos. v. Por insuficiencia en la cantidad de hemoglobina o del hematocrito Debemos tener siempre presente que la anemia es un hecho clínico (signo) y no una entidad diagnóstica (enfermedad), por lo que siempre debemos buscar y tratar el hecho causal.
ETIOLOGÍA DE LA ANEMIA No se trata de una enfermedad sino de la manifestación de un proceso patológico. Causas: v Producción insuficiente de eritrocitos v Perdida excesiva de sangre o hemorragia La Destrucción clasificaciónexcesiva etiológicade deeritrocitos la anemia esta relacionada con las anomalías v clínicas que la) originan: (hemolisis q Hipoproduccion de eritrocitos q Hemorragias agudas o crónicas q Aumento en la destrucción de eritrocitos: hemolisis Intravascular y Extravascular
Clínica. q Cardiovasculares y respiratorios Los síntomas cardiológicos pueden extenderse desde disnea de esfuerzo, taquicardia, hipotensión postural, ángor e infarto de miocardio. • Neurológicos: Cefaleas, acúfenos, vértigo, mareo, pérdida de concentración, astenia, menor tolerancia al frío. q Cutáneos-mucosas: Es típico la palidez de piel y mucosas, siendo en los individuos muy pigmentados la observación de las conjuntivas, lechos ungueales y las líneas de la palma de la mano ü Gastrointestinales: Anorexia, náuseas, estreñimiento o diarrea Ø Genitourinarios. Puede presentar amenorrea. Perdida de la libido, e impotencia.
La anemia ferropénica puede ser debida a tres causas principalmente: Un descenso del aporte de hierro en la dieta. Una disminución de la absorción del hierro a nivel del aparato digestivo por diferentes causas, como pueden ser una menor producción de jugos gástricos (que favorecen la absorción del hierro a nivel intestinal), enfermedades de absorción como por ejemplo la celiaquía, o bien cirugías que afecten al estómago Un aumento de las pérdidas de sangre, como pueden ser sangrados gastrointestinales crónicos, menstruaciones abundantes, otras pérdidas ginecológicas o bien el embarazo, dado que el feto utiliza 2/3 del hierro que absorbe la madre.
Dentro de las pruebas que se pueden realizar tenemos: ü Sideremia: se trata de los valores de hierro plasmático ü Ferritina: es una prueba para ver los depósitos de hierro en el organismo. Se altera en la anemia ferropénica. (bajo) ü Transferrina: es una proteína que se encarga de transportar el hierro en el plasma. Su síntesis se aumenta en la anemia ferropénica. ü Índice de saturación de la Transferrina: indica la capacidad de fijación del hierro a la Transferrina.
Anisocitosis Microcitos Hipocromicos Poiquilocitocis Dianocitos o codocitos MCV disminuidos
ÍNDICES ERITROCITARIOS PARA LA CLASIFICACIÓN DE LAS ANEMIAS
El VCM se puede calcular mediante la fórmula siguiente: VCM = Hematocrito ´ 10 N. de hematíes (´ 1012/L) Este índice eritrocitario permite clasificar las anemias en macrocíticas (VCM > 97 f. L), normocíticas (VCM = 83 -97 f. L) o microcíticas (VCM < 83 f. L). La coexistencia de hematíes de diferentes tamaños se denomina anisocitosis
El contenido hemoglobínico promedio de cada hematíe se expresa por la HCM, que se calcula mediante la siguiente fórmula: HCM = Hb (g/d. L) ´ 10 N. o de hematíes (´ 1012/L) A los hematíes con una HCM disminuida se los denomina hipocrómicos El índice que expresa la concentración de Hb de cada hematíe se conoce como CCMH y se calcula mediante la siguiente fórmula: CCMH = Hb (g/d. L) X 100 Hematócrito (%) la CCMH permanece normal. Es por este motivo que resulta inapropiado hablar de hematíes hipercrómicos. Excepto en situaciones muy concretas, como la esferocitosis hereditaria, la drepanocitosis y la hemoglobinopatía C, la CCMH rara vez supera los 36 g/d. L, valor que está próximo al del límite superior de la solubilidad de la Hb. Mayores concentraciones harían que ésta cristalizara.
ANEMIA MEGALOBLASTICA POR DÉFICIT EN LA MADURACIÓN DEL ERITROCITO La falta de ácido fólico es la causa más frecuente de anemia megaloblástica. El ácido fólico se obtiene de la dieta, a partir de alimentos como la carne, las legumbres, los frutos secos o las verduras, y se absorbe a nivel del yeyuno. Se transporta a la médula ósea para intervenir en la síntesis de ADN de los hematíes y se acumula en depósitos en el hígado. La principal causa de falta de ácido fólico es su déficit en la dieta, sobre todo por desnutrición y alcoholismo, aunque también puede ser debido a alteraciones en su absorción y almacenamiento causadas por diversas enfermedades intestinales y hepáticas, a situaciones que aumenten su consumo, como el embarazo o ciertos tumores, o a algunos fármacos como el metrotexate, el trimetoprim o el triamterene.
La vitamina B 12 se absorbe a nivel del intestino delgado tras unirse en el estómago a una proteína que se llama factor intrínseco. Pasa a la sangre y se transporta unida a otra proteína llamada transcobalamina que la lleva a la médula ósea para intervenir en la formación de los hematíes. La porción que no se usa se almacena en el hígado. Macrocitos Normoblastos Cariorrexis Punteado basófilo Anillos de cabott Cuerpos de howell jolly Test de Shilling
La falta de vitamina B 12 puede ser debida a un déficit de la misma en la dieta, por una ingesta pobre en productos de origen animal, o a un defecto en su absorción intestinal por falta de factor intrínseco, alteraciones en sus receptores, alteraciones de la mucosa intestinal, enfermedades pancreáticas o infecciones intestinales. También puede ser causada por mal uso de la vitamina B 12 o bien por un aumento de las necesidades de la misma, como en el embarazo, por tumores, hipertiroidismo o ciertos fármacos. La atrofia puede ser debida a una enfermedad autoinmune denominada anemia perniciosa, en la cual el organismo crea anticuerpos contra las células de la mucosa, cosa que impide la formación de factor intrínseco y por lo tanto de la absorción de vitamina B 12. Además, al existir una atrofia de la mucosa, se producen menos jugos gástricos, cosa que también dificulta la absorción de hierro, hecho que puede agravar la anemia.
ANEMIA APLASICA Cuando el tejido hematopoyético de la médula ósea desaparece, el espacio queda dentro del entramado óseo es sustituido por tejido graso. Dentro de los diferentes tipos de anemias aplásicas se diferencian las congénitas de las adquiridas. Dentro de las causas de anemia aplásica congénita destacan: anemia de Fanconi, una enfermedad hereditaria autosómica recesiva que además de anemia se presenta con malformaciones cutáneas y óseas; puede llegar a degenerar en una leucemia. Disqueratosis congénita, una enfermedad hereditaria ligada al cromosoma X que asocia aplasia medular y lesiones cutáneas. Síndrome de Diamond Blackfan, una aplasia selectiva congénita que solamente afecta a las células de la línea roja, los hematíes, respetando la línea blanca (leucocitos) y la plaquetaria.
Dentro de las causas adquiridas destacan: fármacos como el cloramfenicol, las sulfamidas, las tiacidas, las sales de oro o la tolbutamida. tóxicos como el benceno, el tolueno, el DDT o el cloruro cálcico virus como el de la hepatitis C, el togavirus, el VIH, los herpes virus o el parvovirus B-19 eritroblastopenia selectiva adquirida, en un 50% de los casos asociada a un tumor del timo. De todos modos, cabe decir que un 50% de las anemias aplásicas adquiridas son de origen desconocido.
Diagnóstico ü El diagnóstico se basará en el interrogatorio del paciente y los datos analíticos: hemograma, bioquímica básica y reticulocitos. ü La prueba de confirmación de la anemia aplásica la proporciona la biopsia de médula ósea, que permitiría estudiar los hematíes en formación así como las otras líneas celulares y se verá que hay un descenso de las células que se forman en ella.
A grandes rasgos, las anemias hemolíticas se clasifican en aquellas causadas por una alteración de los mismos hematíes, llamadas intracorpusculares, y las debidas a alteraciones en los vasos, denominadas extracorpusculares. Las intracorpusculares son congénitas mientras que las extracorpusculares son casi en su totalidad adquiridas.
MEMBRANOPATIAS LA ESFEROCITOSIS HEREDITARIA Está causada por una variedad de mutaciones en los genes que transcriben para la espectrina (el defecto más frecuente), la ankirina (o anquirina), y otras proteínas de la membrana del hematíe. Estas proteínas son necesarias para mantener la forma normal del hematíe, que es la de un disco bicóncavo
Diagnostico § La hemoglobina corpuscular media (HCM) es normal, pero la concentración hemoglobínica corpuscular media (CHCM) está aumentada en el 50 % de los pacientes, lo que refleja una deshidratación de una parte de la población celular. ü La fragilidad osmótica del glóbulo rojo, la cual mide la habilidad de los glóbulos rojos de incrementar su volumen cuando son sometidos a soluciones hipotónicas de cloruro de sodio (Na. Cl) de concentraciones variables. Debido a que los esferocitos tienen una relación superficie/volumen disminuida, tienen una capacidad disminuida para aumentar su volumen y se lisan a una concentración de sales más elevada que las células normales.
q El análisis de las proteínas de la membrana eritrocitaria en electroforesis de poliacrilamida con SDS (PAGE-SDS) se utiliza para cuantificar las proteínas y/o detectar péptidos truncados con migración anormal, y el estudio molecular ha permitido identificar un número importante de mutaciones en distintas proteínas de la membrana eritrocitaria
ELIPTOCITOSIS ü Ocurre como consecuencia de la inestabilidad del esqueleto de membrana debida a la alteración cualitativa o cuantitativa de sus proteínas, que impiden la formación de tetrámeros de espectrina. Así, el eritrocito pierde la capacidad para recuperar su forma tras una deformación longitudinal. La causa más frecuente de EC es la deficiencia de α -espectrina (60%), seguida de la deficiencia de proteína 4. 1 (20 -30%), β -espectrina y glucoforina C. Diagnostico Electroforesis de proteínas de membrana del glóbulo rojo
ENZIMOPATIAS La deficiencia de glucosa 6 -fosfato deshidrogenasa (G 6 PD) es la anomalía enzimática del glóbulo rojo (GR) más frecuente en todo el mundo La G 6 PD participa del ciclo de las pentosas, cuyo objetivo es producir energía como NADPH (Nicotinamida adenina dinucleótido fosfato), para permitir reacciones de oxido-reducción en las diferentes células del organismo. El GR depende exclusivamente de este mecanismo para obtenergía. Al existir la deficiencia enzimática, y en contacto con oxidantes, el hematíe no es capaz de revertir la reacción y se produce la hemólisis Diagnostico Este examen mide la cantidad de glucosa-6 -fosfato dehidrogenasa en la sangre, para evaluar y manejar la deficiencia de la enzima.
La deficiencia de piruvato quinasa se hereda de forma autosómica recesiva. Está causada por mutaciones en el gen PKLR, el gen que codifica el hígado y el tipo de células rojas de la piruvato quinasa. El gen está localizado en el cromosoma 1 (1 q 21). ü Dosificación de la enzima ü Biología molecular
HEMOGLOBINOPATÍAS Son enfermedades hereditarias de la sangre que alteran el transporte de oxígeno. Se dividen en dos categorías principales: la drepanocitosis y las talasemias.
TALASEMIAS Son anomalías cuantitativas en la síntesis de las diferentes cadenas que integran la hemoglobina. • Las hemoglobinas anormales son el resultado de la eliminación, adición o sustitución de uno o mas aminoácidos por otro u otros en las cadenas polpeptidicas que las componen o de la fusión de estas. • Las talasemias conservan la composición estructural de la molécula de hemoglobina, pero existe una síntesis defectuosa de las cadenas globinicas del tetrámero de la hemoglobina. v Talasemia mayor: hay disminución de las cadenas beta de Hb, disminución de la Hb. A 1 y amento de la Hb. A 2 y aumento de la Hb. F, aumento en la absorción del hierro, hiperplasia en la medula ósea y malformaciones óseas. v Talasemia menor: hay disminución del VCM, y alteración en el % de Hb. A 2>, <Hb. A 1
Hemoglobinopatía S Anemia de células falciformes consiste en una sustitución de la cadena beta de acido glutámico en la posición 6 por una molécula de valina. Estos hematíes colapsan la microcirculación sanguínea, ocasionando las denominadas crisis vasooclusivas, que producen isquemias de órganos múltiples y en situación prolongada infartos. Además la deformabilidad del glóbulo rojo ocasiona la destrucción del mismo y por tanto su hemolisis es intravascular. Diagnostico prueba de ciclaje y la electroforesis de hemoglobina.
ESP Talasemia ESP Drepanocitosis
Anemias hemolíticas adquiridas § Hiperesplenismo suele asociarse a la destrucción de células hematológicas en el bazo o secuestro esplénico. § Hemolisis química arsénico, cobre, anfoterixina B, venenos de arañas, serpientes, toxinas bacterianas Clostridium, estos producen lesión directa en la membrana del hematíe. § Alteraciones metabólicas hiperlipoproteinemias y hepatopatías que alteran los lípidos plasmáticos. § Parasitosis: malaria y babesiosis, filarias § Trauma eritrocitario: hay presencia de esquiztocitos en sangre periférica. § Hemoglobinuria de la marcha: se produce hemolisis intravascular como consecuencia de traumatismos repetidos al caminar. § Patología cardiovascular: estenosis o insuficiencia aortica, bypass y prótesis valvulares. § Alteración en la microcirculación: anemia microangiopatica CID, hemangiomas, rechazo de injerto renal, hipertensión maligna, eclampsia, vasculitis, neoplasias diseminadas, SHI, Purpura trombocitopenia trombotica.
Ø ANEMIAS INMUNOHEMOLITICAS Ø Se denomina así ya que la Complemento. hemolisis mediada por inmunoglobulinas y/o Ø Las Inmunoglobulinas. Pueden ir dirigidas contra Ag. Extraños como ocurre en las reacciones postransfusionales o la enfermedad hemolítica del recién nacido o bien ser anticuerpos que reaccionan con antígenos eritrocitarios propios como consecuencia directa de agentes externos que modifican los antígenos del hematíe. Ø La inmunohemolisis se produce por la activación del complemento habitualmente por la Ig. M. Y a veces por Ig. G. Ø Se produce una destrucción inmediata de la membrana del hematíe ya que las ultimas fracciones del complemento C 5 -C 9 tienen acción lítica sobre la membrana. Ø Como la mayor parte de los hematíes se encuentran en circulación sanguínea la hemolisis mediada por el complemento suele ser predominantemente intravascular.
v La hemolisis que no es mediada por el complemento sino por Inmunoglobulinas. Habitualmente Ig. G. v Tiene su lugar fundamental de activación en el bazo ya que los macrófagos esplénicos presentan receptores de membrana para la fracción constante de la Ig. G. v La captación del hematíe por el macrófago da lugar a destrucción completa o bien parcial, produciendo una lisis de la membrana del hematíe por la fagocitosis. Hemolisis extravascular Prueba de- Bazo laboratorio diagnostica es el Coombs v Dicha prueba puede detectar Inmunoglobulinas complemento sobre la membrana del eritrocito. (CD) v Detectar anticuerpos en el plasma (CIND) O
Anemias caracterizadas por hemólisis intravascular Ig. M Activación del complemento en la membrana del eritrocito v. Hemoglobinuria paroxística nocturna v. Hemoglobinuria paroxística por frío v. Algunas anemias hemolíticas autoinmunes v. Anticuerpos fríos 20 -30% anemias inmuhemoliticas Traumatismos físico o mecánico del eritrocito üAnemia hemolítica microangiopática üAnormalidades del corazón y los grandes vasos üCoagulación intravascular diseminada Microambiente tóxico q. Infecciones bacterianas q. Infección por P. falciparum q. Venenos q. Etc Anemias caracterizadas por hemólisis extravascular Ig. G Defectos hereditarios del eritrocito o. Talasemia o. Hemoglobinopatías o. Trastornos de la membrana Defectos adquiridos del eritrocito • Anemia megaloblástica • Anemia de células en espuela • Deficiencia de vit. E en recién nacidos Anemias inmunohemolíticas ØAutoinmunitaria ØAnticuerpos calientes 70% de las anemias inmunoheloticas común en mujeres ØInducida por fármacos
Hemoglobinuria paroxística nocturna ü Aunque la enfermedad cursa con anemia hemolítica, se trata de un proceso mucho más complejo, ya que es un trastorno de la célula madre pluripotencial de la médula ósea (procesos denominados panmielopatías clonales). ü Las células derivadas de esta célula madre anormal tienen como característica un exceso de sensibilidad al complemento. ü Dado que se afectan las tres series hematológicas, es frecuente la presencia de pancitopenia. ü Las células hematológicas son sensibles al complemento porque carecen de la presencia de sustancias que inhiben la acción del complemento sobre la membrana del hematíe (CD 55 y CD 59). ü Al faltar dicha sustancia, pequeñas activaciones del complemento, aun fisiológicas, pueden ocasionar destrucción de la membrana, no sólo de los hematíes, sino también de los leucocitos y las plaquetas. ü Dado que se trata de una hemólisis mediada por el complemento, si esta hemólisis es severa se acompaña de hemoglobinuria.
Mieloptísica anemia q Es un tipo grave de anemia que se encuentra en algunas personas con enfermedades que afectan a la médula ósea. q La Myelophythisis se refiere al desplazamiento de tejido de médula ósea hematopoyéticas en la sangre periférica, ya sea por la fibrosis, tumores o granulomas. Causas v La Myelophythisis puede ocurrir en el contexto de una enfermedad crónica. v De una enfermedad mieloproliferativa, la leucemia, el linfoma, y el carcinoma metastásico o mieloma. v Es común en las personas que tienen la mielofibrosis idiopática crónica. v Se ha relacionado con el cáncer de pulmón de células pequeñas, cáncer de mama o cáncer de próstata que hace metástasis a la médula ósea.
Diagnóstico ü La primera prueba para mielotisis diagnóstico consiste en examinar una pequeña muestra de sangre bajo el microscopio. ü Mielotisis se sugiere por la presencia de células rojas de la sangre que contienen núcleos o son en forma de lágrima
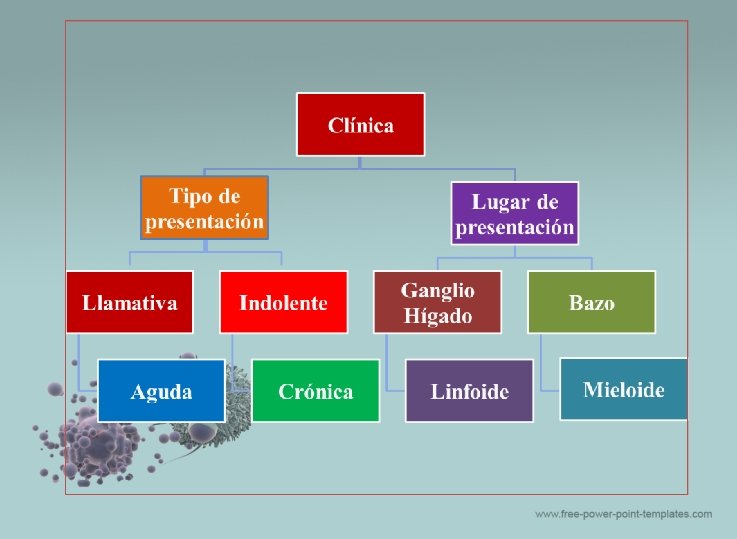
LEUCEMIAS Consiste en la proliferación incontrolada de una población anómala de células de la sangre. Estas células anómalas infiltran la médula ósea, impidiendo la producción de las restantes células normales, e invaden la sangre y otros órganos.
CLASIFICACIÓN q Leucemia linfoide agudas crónica v Leucemia mieloide aguda crónica Agudas Crónicas
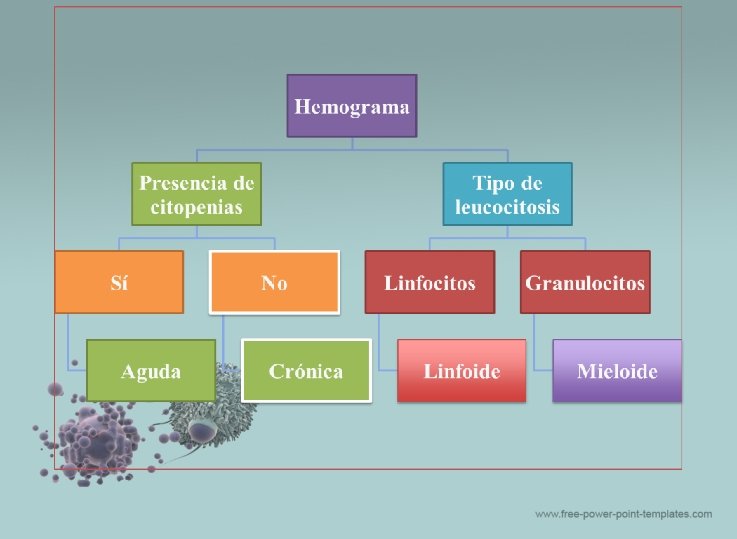
Leucemias Agudas ü Son patologías que surgen debido a la proliferación descontrolada de las stem cells hematopoyéticas las cuales son incapaces de madurar adecuadamente, por lo tanto se detienen en fases tempranas de la maduración observándose blastos en cantidades variables en sangre periférica , los cuales invaden la medula ósea y pueden producir focos extra hemáticos comportándose como tumores. ü Debido a la invasión de la medula ósea , los demás componentes celulares disminuyen en su producción, presentándose generalmente anemia, trombocitopenia y neutropenia.
1) Tipo Clínica Hemograma Leucemia linfoide aguda Fatiga –Fiebre Sangrado, infecc. Adenopatías Dolores –SNC Anemia Trombocitopenia Granulocitopenia Linfocitosis Leucemia Mieloide aguda Fatiga –Fiebre Sangrado –esplenomegalia Dolores óseos Anemia Leu 15, 000 -5000 Plaq <100, 000 Leucemia linfoide crónica Adenopatías Esplenomegalia mina Hepatomegalia Linfocitosis (10, 000) Anemia 15% Trombopenia 15% Leucemia mieloide crónica Infecciones, hemorragia, trombosis Esplenomegalia masiva Tumores en diferentes ubicaciones. Leu >100, 000 Granulocitosis trombocitosis
LEUCEMIA LINFOIDE AGUDA Enfermedad caracterizada por la proliferación clonal no controlada de células linfoides inmaduras de linaje B o T. Afecta a las células de la estirpe Linfoide y son las más frecuentes en los niños. L 1, L 2, L 3 L 1: Leucemia Linfática Aguda o Linfoblastica típica: es la más común dentro de las Leucemias Linfáticas en niños, se encuentran blastos pequeños, escaso citoplasma, nucléolos poco visibles y cromatina homogénea.
L 2: Leucemia Linfática Aguda o Linfoblastica atípica: más frecuente en adultos. Presenta dos tipos de blastos: unos grandes, núcleo irregular, con uno a dos nucléolos visibles, citoplasma con basófilo variable y otros que son pequeños parecidos a los L 1. L 3: Leucemia Linfoblastica Aguda o tipo Burkitt: es de muy baja frecuencia, los blastos son característicos ya que poseen un citoplasma muy basófilo con vacuolas abundantes, núcleo grande con nucléolos visibles.
Leucemia Mieloide Aguda Para clasificar una Leucemia como mieloide se deben tener en cuenta los parámetros expuestos previamente como lo es la inmunofenotipificacion la cual nos entregara información acerca de la presencia de marcadores de linaje mieloide y la morfología en la cual se debe describir las características de los blastos que se encuentran en el hemograma fijándose en 4 aspectos: • Tamaño o tamaños • Relación Núcleo/citoplasma: alta, Baja, intermedia • Núcleo: con o sin nucléolos, forma, cromatina (laxa o densa) • Citoplasma: tamaño, forma, basofilia, inclusiones, granularidad Las Leucemias mieloides Agudas según la FAB se dividen en 8 variedades por lo tanto van de la M 0 a la M 7: • M 0: Leucemia Mieloide Aguda Indiferenciada: son células muy inmaduras las cuales no expresan morfológicamente ningún elemento que las clasifique como Mieloide (gránulos citoplasmáticos, Bastones de Auer) y solo se encuentran aquí debido a que su Inmunofenotipo corresponde al de la estirpe mieloide (CD 13+, CD 33+). Blastos: citoplasma agranular, núcleo con cromatina laxa con 1 -2 nucléolos
• M 1: Leucemia Mieloide Aguda sin maduración: blastos con más maduración que M 0, tienen núcleo con cromatina Laxa, con 1 -2 nucléolos, citoplasma con finas granulaciones, relación núcleo citoplasma alta y presencia de bastones de Auer en un pequeño porcentaje. • M 2: Leucemia Mieloide Aguda Diferenciada o con Maduración: Blastos con signos visibles de linaje mieloide, núcleo irregular con nucléolos, cromatina laxa, citoplasma abundante granular que puede presentar bastones de Auer • M 3: Leucemia Mieloide Aguda Promielocitica: en esta leucemia se observan blastos y promielocitos en alta cantidad los cuales presentan un citoplasma hipergranular, núcleo irregular generalmente lobulado, presencia de Bastones de Auer en gran cantidad (en empalizada). Asociada a la translocación t(15, 17) la cual es de buen pronóstico. Posee una variante Hipogranular donde se observan promielocitos sin gránulos y sin bastones de Auer. • M 4: Leucemia Mieloide Aguda Mielo. Monocitica: Se observan 2 tipos de blastos uno son grandes de forma irregular y los otros son medianos y redondos. Comparten marcadores tanto de la serie mieloide como monocitoides. Poseen una variante en la cual se observan Eosinófilos en abundancia.
M 5: Leucemia Mieloide Aguda Monocitica: se divide en 2 variedades: M 5 a-> presencia de monoblastos de gran tamaño con marcadores mieloides y monocitoides. M 5 b-> Presencia de monocitos irregulares con núcleo arriñonado o plegado, citoplasma azul, basófilo y finas granulaciones. Posee marcadores monocitoides. M 6: Eritroleucemia: La serie eritroide corresponde a más del 50% de las células nucleadas y los blastos son más del 20%. El eritroblasto es de forma circular con núcleo regular, citoplasma muy basófilo con un halo blanco. Presentan además depósitos anormales de hemosiderina (observación con tinción para Hemosiderina). M 7: Megacarioblastica: presencia de megacarioblastos en la sangre periférica y restos de núcleo de megacariocito. Son células de formas y tamaños irregulares de núcleos con cromatina laxa.
Bastones de auer Consisten en material granulado, compuesto de lisosomas fusionados que contienen peroxidasa, enzimas lisosomales y cristales, Por lo general, aparecen característicamente en mieloblastos de la leucemia mieloide aguda, en especial las de tipo M 1, M 2, M 3 y M 4.
Leucemias Crónicas Son alteraciones neoplásicas caracterizadas por la proliferación descontrolada de los precursores medulares, sin embargo a diferencia de las leucemias agudas, en las leucemias crónicas se producen alteraciones en etapas avanzadas de la maduración observándose en sangre periférica todos los estadios de maduración correspondientes a la línea celular afectada. Estas Leucemias se encuentran dentro de los síndromes Linfoproliferativos y Mieloproliferativos según la línea celular afectada. Leucemia Mieloide Crónica Corresponde a un síndrome mieloproliferativo que es más común en pacientes de la tercera edad, el cual se encuentra asociado en un alto porcentaje a la traslocación de los cromosomas 9 y 22 t (9, 22) generándose un cromosoma pequeño (cromosoma Philadelphia) y produciéndose una proteína quimérica BCR/ABL la cual induce alteraciones en el control de la proliferación y apoptosis celular de la serie granulocitica.
Posee 3 fases de evolución en las cuales la anemia, trombocitopenia y blastos se hacen más visibles: Ø Fase Crónica: caracterizada por anemia leve normo citica/normo crómica, Recuento de leucocitos mayor a 100, 000 /mm 3 (generalmente recuentos de 400, 000), trombocitosis leve y presencia de macroplaquetas y con una formula diferencial en la cual se observa toda la línea granulocitica. Ø Fase Acelerada: caracterizada por anemia normo crómica con anisocitosis leve, recuento de leucocitos mayor a 100, 000/ mm 3, Formula diferencial en la cual se observa un aumento del porcentaje de blastos, Recuento plaquetario normal. Ø Fase Blastica: Anemia moderada con anisocitosis, anisocromia y poiquilocitosis, un recuento de leucocitos disminuido, normal o aumentado, formula diferencial con un
Leucemia Linfática Crónica ü Corresponde a un síndrome Linfoproliferativo caracterizado por presentar un alto % de linfocitos en sangre periférica y medula ósea. Los linfocitos son morfológicamente normales de tamaño pequeño con escaso citoplasma y de aspecto maduro. ü El recuento de leucocitos esta aumentado y no se presenta con anemia. Además en el frotis se observan las sombras de Gumprecht las que corresponden a restos celulares que se observan en alta cantidad.
Tricoleucemia o Leucemia de Células Velludas ü Corresponde también a un síndrome Linfoproliferativo que afecta a los linfocitos B, el cual se da principalmente en hombres de 40 años, cuyos síntomas principales son infecciones recurrentes y sangrado de encías en conjunto con esplenomegalia. ü En el frotis se observan células muy características de tamaño variable, núcleo con cromatina laxa y un citoplasma con prolongaciones. ü Debido a su morfología es que a estas células se les conoce como velludas y de ahí el nombre Leucemia de las células Velludas
Diagnóstico: o Para diagnosticar una Leucemia Aguda se necesitan varios exámenes que lo confirmen: o Hemograma: hallazgo de más de un 20% de blastos en sangre periférica, anemia, trombocitopenia, leucocitosis ->Morfología de las células describiendo las características del núcleo y del citoplasma teniendo como base la hematopoyesis normal. o Exámenes de medula ósea: Mielograma y Biopsia Medular o Inmunofenotipo: determina el linaje de las células neoplásicas (mieloblasto, linfocito, etc. ) a través de marcadores de superficie los cuales son detectados a través de la citometria de flujo o Citogenética y Biología Molecular: busca las posibles alteraciones genéticas y cariotipicas que se asocian al pronóstico de la patología que presenta el paciente. o Tinciones Citoquímicas (solo en ocasiones).
GAMATIAS MONOCLONALES v son un grupo diverso de trastornos caracterizados por una clona (células genéticamente idénticas) de linfocitos y células plasmáticas, con capacidad de producir una inmunoglobulina o un fragmento de la misma, que puede detectarse en sangre y/u orina en forma de una banda o componente monoclonal. q QUÉ ES UN MIELOMA MÚLTIPLE Existen otras células como las células plasmáticas, que derivan de un tipo de glóbulo blanco llamado linfocito B y producen anticuerpos, sustancias que sirven para defender al organismo de infecciones y otras agresiones externas. El mieloma múltiple es un cáncer producido por la acumulación anormal de gran cantidad de células plasmáticas en la médula ósea. Cuando las células plasmáticas se multiplican de forma descontrolada pueden provocar: • Una reducción del número de células madre en la sangre. • La producción defectuosa y abundante de anticuerpos que resultan inservibles. • Existencia en la sangre y/o en la orina de una sustancia llamada proteína M o monoclonal, que puede dañar el riñón. • La activación de los osteoclastos, que son unas células que destruyen el hueso.
Fenómeno de roleaux Células plasmáticas Diagnostico mieloma múltiple ü Mas de un 30% de células plasmáticas en M. O ü Pico monoclonal en electroforesis de proteínas séricas Ig. G >3, 5 g/ml, ü Ig. A>2 g/ml. ü Lesiones líticas o fracturas patológicas de hueso largos ü Calcio sérico >12 mg/dl ü Creatinina sérica >2 mg/dl
Síndromes mielodisplasicos Son un grupo de enfermedades clónales de la célula madre hematopoyética caracterizada por citopenias, displasia anomalía morfológica, en una o mas líneas celulares, hematopoyesis ineficaz y un riesgo elevado de desarrollar leucemia mieloide aguda Aspectos esenciales - Hay que sospechar de un síndrome mielodisplasico ante un anciano con anemia y VCM normal o elevado. - La mayoría de SMD son idiopáticos suelen asociarse a quimioterapia y radioterapia. - Clínicamente producen citopenias (anemia, infección, hemorragia), pudiendo evolucionar a leucemia aguda con exceso de blastos. - La anemia que se produce es hiporregenerativa (reticulopenia). - La medula ósea es hipercelular. - El trasplante de MO no es posible porque casi siempre es ancianos.
v Sangre periférica v Serie roja: anemia normocitica o macrocitica con reticulocitos disminuidos. v Serie blanca: hay leucopenia, alteraciones de los leucocitos (leucocitos hipogranulares, o anomalía de pseudopelger, déficit enzimático de la fosfatasa alcalina leucocitaria, al igual que en LMC. v Serie plaquetaria habitualmente hay trombocitopenia con anisotrombia y disfuncionalidad plaquetaria. v Existe una variante de SMD asociado a un trastorno citogenetico que es la delecion parcial del brazo largo del cromosoma 5 (síndrome 5 -q) Clasificación ü Anemia refractaria simple , alteración en la biosíntesis del hemo, que afecta a la serie roja blastos<5%. ü Anemia refractaria con exceso de blastos, el % de blastos medulares oscila entre 5% y 20% ü Anemia refractaria con exceso de blastos en transformación, el % de blastos medulares oscila entre 20% y un 30%. ü Anemia refractaria con sideroblastos en anillo, esta enfermedad se considera cuando existe mas de un 15% de precursores de la serie roja con hierro de deposito en forma anular alrededor (sideroblastos en anillo)
En el linfoma de Hodgkin (LH) se producen de forma descontrolada unas células linfoides atípicas, denominadas de Reed-Sternberg, que parecen originadas en los linfocitos B, causando el aumento de tamaño de los ganglios de una región del organismo para extenderse con el tiempo a otras áreas ganglionares vecinas, al bazo o la médula ósea.
v En los LNH, una célula linfoide, detenida en un determinado estadio madurativo, se reproduce de forma incontrolada, causando con el tiempo el aumento de tamaño del órgano en el que se producen. v Dado que el tejido linfático se encuentra en todo el cuerpo, los linfomas pueden aparecer en cualquier parte del organismo y, a partir de allí, diseminarse a otros órganos y tejidos. v La mayoría de los casos empiezan con una infiltración en un ganglio linfático (formas nodales), pero algunos subtipos específicos pueden estar restringidos a la piel, cerebro, bazo, corazón, riñón u otros órganos (formas extranodales). 159
REPORTE DE UNA CÉLULA PROBLEMA