Seminar on Methods to Enhance Dissolution Rate Contents

















 The α-, β-, γ- cyclodextrins are")













- Slides: 33
Seminar on Methods to Enhance Dissolution Rate
Contents Ø Introduction Ø Process of dissolution Ø Methods for enhancing dissolution rate Ø Conclusion Ø References
Introduction Dissolution is the process by which a solid substance solubilizes in a given solvent. Dissolution rate is defined as the amount of solid substance that reaches into solution per unit time under standard conditions of temperature, pressure, and p. H. Concept of dissolution was introduced in 1897 by Noyes and Whitney, and also they had published a paper on “Utilizing cylindrical sticks of lead chloride and benzoic acid, and they tried to measure the rate at which a sparingly soluble substance, with a constant surface area. They suggested that dissolution rate is controlled by the rate of diffusion of a very thin layer of saturated solution that forms instantaneously around the solid. Noyes and Whitney have proposed an equation
Process of dissolution The process of dissolution involves breaking of inter-ionic or intermolecular bonds in the solute, the separation of the molecules of the solvent to provide space in the solvent for the solute, and the interaction between the solvent and solute molecule or ion.
Methods to Enhance Dissolution Rate 1. 2. 3. 4. 5. 6. 7. 8. Increase in the effective surface area of the drug. Particle size reduction Incorporation of surface active agents in formulation. Solute-Solvent complexation reactions. Polymorphism. Molecular encapsulation with cyclodextrins or complexation with cyclodextrins. Prodrug approach. Salt formation of drug.
1. Increasing the effective surface area of the drug The size of the solid particle influences the solubility because as particle becomes smaller, the surface area to volume ratio increases. The larger surface area allows a greater interaction with the solvent. The effect of particle size on solubility can be described by Where So is the solubility of infinitely large particles and S is solubility of fine particles, V is molar volume, γ is surface tension of solid and r is the radius of fine particle.
2. Particle size reduction Micronization technique: Increasing dissolution by reducing particle size of poorly water soluble drugs. Reduction is done by variety of micronization process such as grinding, ball milling, air attrition, utilization fluid energy mills. Micronization process usually results in 1 -10µm diameter. Kronblum and Hirschorn evaluated two specific methods of micronization, spray drying and air attrition, which provided drug forms of different specific surface areas and particle size ranges, as well as other physical characteristics.
3. Incorporation of Surface Active Agents This helps by enhancing dissolution by increasing wetting and penetration properties of the dissolution medium through incorporation of surface-active agent. Concentration usually employed is below the CMC(critical micellar concentration) value which is insufficient to cause any increase in solubility but increases the drug surface area exposed to the medium by lowering the surface tension of the dissolution medium. Usually non-ionic surfactants are used in formulating insoluble drugs in the form of soft elastic capsules.
• Surfactants can also be used to enhance solubility. A surfactant or surface active agent is amphipathic, meaning it has polar end (the circular head) and a nonpolar (the tail). When a surfactant is placed in water it will form micelles. A nonpolar drug will partition into the hydrophobic core of the micelle and the polar tails will solubilize the complex.
4. Solute- Solvent Complexation Reactions Molecular complexation between molecules of dissolving solutes and certain solvents have been known to effect dissolution rates. Higuchi studied the dissolution rate of 2 -napthol tablets in cyclo hexane containing various amounts of 1 -propanol and 1 -undecenol. These additives are known to react rapidly and reversibly forming soluble complexes. Major complexation is hydrogen bonding. This can be called as pseudo polymorphism. Dissolution kinetics are controlled by factors like diffusion coefficient of the complexing component of the solvent and the stability constant of the resulting complex. Solute solvent complexation reactions also improve dissolution by reducing particle size of solid.
There are three means by which the particle size can be reduced to sub micron level are: 1. 2. 3. Solid solutions, Eutectic mixtures, and Solid dispersions. In all these cases the solute is frequently a poorly water soluble drug acting as a guest and the solvent is a highly water soluble compound or polymer acting as a host or a carrier.
Solid solutions It is a binary system comprising a solid solute molecularly dispersed in a solid solvent. Since the two compartments crystallize together in a homogenous one phase system, solid solutions are also called as molecular dispersions or mixed crystals. By the reduction in particle size to the molecular level, solid solutions show greater aqueous solubility and faster dissolution. Usually they are prepared by fusion method where by a physical mixture of solute and solvent are melted together followed by rapid solidification. Eg: Griseofulvin-succinic acid Solid solution of Griseofulvin dissolves 6 to 7 times faster than pure griseofulvin.
The two mechanisms suggested for enhanced solubility and rapid dissolution of molecular dispersions are: 1. When the binary mixture is exposed to water is, the soluble carrier dissolves leaving the insoluble drug in a state of microcrystalline dispersion of very fine particles, and 2. When the solid solution, which is said to be in a state of randomly arranged solute and solvent molecules in the crystalline lattice, is exposed to the dissolution fluid, the soluble carrier dissolves rapidly leaving the insoluble drug standard at almost molecular level.
Eutectic Mixtures These systems are also prepared by fusion method. Eutectic melts form solid solutions in that the fused melt of solute-solvent show complete miscibility but negligible solid-solid solubility i. e. , such systems basically intimately blended physical mixture of two crystalline components. When the eutectic mixture is exposed to water, the soluble carrier dissolves leaving the drug in a microcrystalline state which solubilizes rapidly. Examples of eutectic include paracetamol-urea, griseofulvin- succinic acid, etc. Solid solutions and eutectics, which are basically melts, are easy to prepare and economical with no solvents involved. Method cannot be applied to Ø Ø Ø Drugs which fail to crystallize from mixed melt, Thermo labile drugs, and Carriers such as succinic acid that decomposes at their melting point.
Solid Dispersions These are generally prepared by solvent or co-precipitation method where by both the guest solute and the solid carrier solvent are dissolved in a common volatile solvent like alcohol. The liquid solvent is removed by evaporation under reduced pressure or by freeze drying which results in amorphous precipitation of guest in a crystalline carrier. Thus the basic difference between the solid dispersions and solid solutions/eutectics is that the drug is precipitated out later; e. g. : amorphous sulfathiazole in crystalline urea. Such dispersions are often called as Co-evaporates or Co-precipitates. The method is suitable for thermo labile substances. The carriers used are same as for eutectics and solid solutions. With glassy materials, the dispersions formed are called as glass dispersions or glass suspensions.
5. Polymorphism A solid has a rigid form and a definite shape. The shape or habit of a crystal of a given substance may vary but the angles between the faces are always constant. A crystal is made up atoms, ions, or molecules in a regular geometric arrangement or lattice constantly repeated in three dimensions. This repeating pattern is known as the unit cell. The capacity for a substance to exhibit in more than one crystalline form is polymorphism. If the change from one form to another is reversible, the process is called enantiotropic. Use of metastable forms help in increasing the dissolution rate.
The use of Metastable Polymorphs: The solid state characteristics of drug are known to potentially exert the significant influence on the solubility parameter. As the presence of metastable, polymorphic crystalline forms can exert a great influence on solubility, dissolution rate and biological activity of medicaments. The separation and use of specific polymorphic form that possesses the highest solubility is a technique that can be applied in certain cases for the increase of dissolution rates. Drugs which show metastability include Chloramphenicol, Prednisolone, Barbiturates, and Riboflavin.
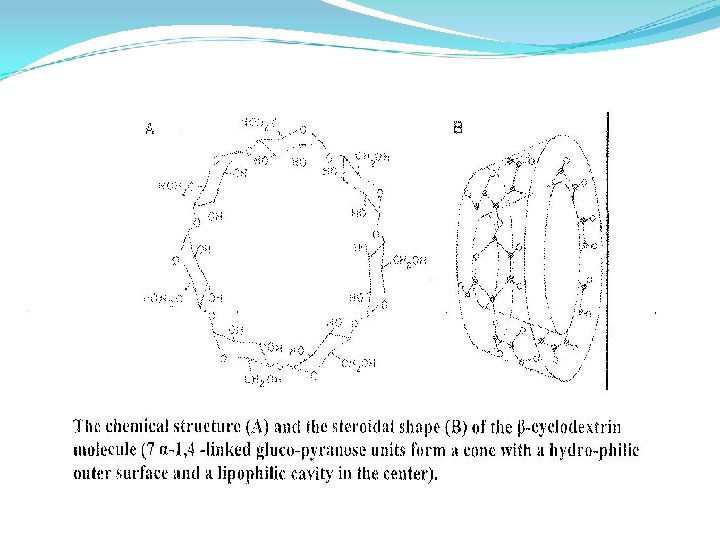
6. Molecular Encapsulation with Cyclodextrins (Complexation with Cyclodextrins) The α-, β-, γ- cyclodextrins are cyclic oligosaccharides consisting of six , seven and eight glucose units respectively. Their important property is ability of forming inclusion complexes with smaller molecules which fit into their hydrophobic cavity of the cyclodextrins. The formation of inclusion complex alters a variety of physico-chemical properties of the drug molecules such as its solubility, dissolution rate, membrane permeability, chemical reactivity and dissociation constant. Solubility increases with increase in the amount of cyclodextrin added. Among the natural cyclodextrins β-cyclodextrin (βCD) is used widely because of its unique cavity size (internal diameter about 6. 5 °A). In 1975, Kurozumi devised a simple freeze drying process for the formation of βCD complexes in the solid state which simulated more interest in this technique for increasing the solubility and dissolution rate of water insoluble drugs.
Marketed preparations Alfaxalone Steroid anesthetic Incorporation : 100 mg/g CD used: THPB Trappsol Hydroxypropyl Beta Erythromycin 1 Antibiotic Incorporation: 45 mg/g CD used: THPB Hydrocortisone Glucocorticoid Incorporation: 90 mg/g CD used: THPB Ibuprofen Anti-inflammatory Incorporation: 90 mg/g CD used: THPB
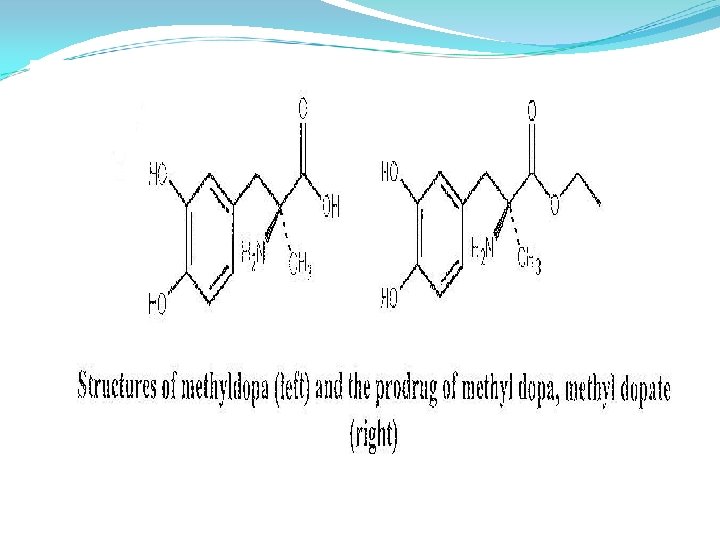
7. Prodrug Approach One method to increase the solubility of a drug is to alter the structure of the molecule. The addition of polar groups like carboxylic acids, ketones and amines can increase solubility by increasing hydrogen bonding and interaction with water. Another structure modification may be can be to reduce intermolecular forces. E. g. : methyldopa (solubility ~10 mg/ml) and methyldopate (10 -300 mg/ml depending on p. H). The addition of ethyl ester to methyldopa reduces the intermolecular hydrogen bond between the carboxylic acid and primary amine. There fore this addition reduces melting point and increases solubility.
Another method to increase solubility is the use of co-solvents. Some commonly used co-solvents in pharmaceutics are propylene glycol, polyethylene glycol, ethanol and sorbitol. The addition of co-solvent can increase solubility of hydrophobic molecules by reducing the dielectric constant of the solvent. The use of co-solvents can increase the solubility by several orders of magnitude. Some problems with the use of co-solvents are precipitation of drug with dilution of solvent mixture and tissue damage or pain upon injection. This dilution occurs after administration of drug in the body.
• Final method is complexation. Complexation lies on relatively weak forces such as London forces, hydrogen bonding and hydrophobic interactions. Type Example Coordination Hexamine cobalt (III) chloride Chelates EDTA Metal-Olefin Ferrocene Inclusion Choleic acid Molecular complexes Polymers
8. Salt form of the drug Most of the drugs are either weak acids or weak bases. One of the easiest method to enhance dissolution rate of drugs is to convert them into salt forms. At a given p. H, the solubility of a drug, whether acidic/basic or its salt form is a constant. The influence of salt formation on the drug solubility, rate of dissolution and the absorption can be explained by considering the p. H of the diffusion layer and not the p. H of the bulk of the solution. Consider a case of a salt of weak acid at any given p. H of the diffusion layer of the salt of the weak acid will be higher than that observable with the free acid from the drug.
Newer techniques Self emulsification Self emulsifying agent will act as dispersing or self emulsifying agent on drug through which the dissolution of the drug can be increased by preventing the formation of any water insoluble surface layer, although the liberated drug remain undissolved in the dissolution medium. When its concentration exceed its saturation solubility, it will disperse or emulsify into a finely divided state because of surface activity of the dissolved vehicle the surface area will be made available which facilitate its dissolution in gastrointestinal fluid.
Nanosuspensions A nanosuspension is a submicron colloidal dispersion of drug particles which are stabilized by surfactants. The poor water solubility of drugs is major problem for drug formulation. To date, nanoscale systems for drug delivery have gained much interest as a way to improve the solubility problems. The reduction of drug particles into the sub-micron range leads to a significant increase in the dissolution rate and therefore enhances bioavailability. Nanosuspensions are promising candidates that can be used for enhancing the dissolution of poorly water soluble drugs. Nanosuspensions contain submicron colloidal dispersion of pharmaceutical active ingredient particles in a liquid phase stabilized by surfactants. Production of drugs as nanosuspensions has been developed for drug delivery systems as an oral formulation and non-oral administration.
Ternary systems Hydrophilic polymers have been commonly used as carriers for preparing solid dispersions. Among them, Polyvinylpyrrolidone (PVP) was widely employed for its high aqueous solubility, high physiological tolerance, and low toxicity. In recent years, the interest in incorporating a surface-active carrier into solid dispersion increased greatly and a high improvement in drug dissolution was reported. Mura et al reported that the dissolution of naproxen from solid dispersions in polyethylene glycol (PEG) 4000, 6000, and 20000 could be further enhanced when Polysorbate 80 was incorporated into the system. Dannenfelser et al found that a combined carrier consisting of PEG and Polysorbate 80 could improve the dissolution and enhance the bioavailability of LAB 687, a poorly water-soluble drug with an aqueous solubility of 0. 17 μg/m. L at room temperature.
The dissolution profiles of OA, physical mixture, and ternary solid dispersions.
Conclusion For any drug to show proper efficacy and safety it should reach the systemic circulation showing optimum bioavailability that further depends upon the dissolution of the drug dosage form in vivo and this dissolution should be occurring at a required rate. The dissolution can be enhanced to improve the bioavailability. Using proper surfactants, increasing the surface area by reducing the particle size, etc, can enhance the dissolution and the above discussed parameters, thus improving bioavailability and therapeutic efficacy of the medicament. Dissolution depends on the chemistry of the active ingredients and physicochemical properties of the excipients used. Thus by altering physicochemical properties of the excipients such as addition of surfactants, altering the p. H of the microenvironment of the drug, etc so as to achieve the ultimate bioavailability.
References 1. 2. 3. 4. 5. 6. 7. 8. D. M. Brahmankar, Sunil B Jaiswal. Biopharmaceutics and pharmacokinetics 2005; pg no. 29, 290 -296. Abdou. Dissolution of pharmaceutical drugs 2001; pg no. 5, 56 -68. V. Venkateshwar Rao “Biopharmaceutics and pharmacokinetics 2005. Connors KA. The stability of cyclodextrin complexes in the solution. Chem Rev. 1997; 97: 1325 -1357. Uekema K, Hirayama F, Irie T. Cyclodextrin drug carrier systems. Chem Rev. 1998; 98: 2045 -2076. Stella VJ, Rajiwski RA. Cyclodextrins: Their further in drug formulation and delivery. Pharm Res. 1997; 14: 556 -567. Erden N, Celebi N. A study of inclusion complex of naproxen with βcyclodextrin. Int J Pharm. 1988; 48: 83 -89. School of Pharmacy, Walailak University, Nakhon Si Thammarat 80161, Thailand Nanosuspension Technology for Drug Delivery. Walailak J Sci & Tech 2007; 4(2): 139 -153.
Thank You