Semeiotica genetica Vincenzo Nigro Dipartimento di Patologia Generale














 •")
 con pattern")






 \"addormentato\", il più")




")

 plagiocefalia = asimmetria del cranio Frontale o posteriore (posturale? ) • Può essere")
 Cresta metopica = prominenza al centro della fronte fino alla trigonocefalia • È")


 del")

 Macrocefalia • È comunemente non patologica, specie se condivisa in famiglia • Può")
 arti corti e testa sproporzionatamente più")
 che")

 il 15 -20% di")

 sul braccio lungo del cromosoma X")




 Microcefalia • È comunemente autosomica recessiva, ma può essere dovuta a sostanze")
 Varianti della linea di attaccatura dei capelli • Ciuffo ribelle (cowlick) •")
 Turner 1: 2. 500 • Solo l’ 1% delle")
 Turner 1: 2. 500 – mandibola più piccola (micrognazia)")
 Ipertelorismo = occhi più distanziati maggiore sviluppo relativo delle ali dello sfenoide, per")

")
 Ipotelorismo = occhi più vicini Ridotta distanza interpupillare, spesso associato a sindromi cromosomiche")
")
 sinofri = sopracciglia che si incontrano sulla linea mediana È spesso associato ad")
 Micrognazia = mandibola più MANDIBOLA piccola Si vede meglio di profilo a bocca")
 Turner 1: 2. 500 – mandibola più piccola (micrognazia)")
 Asimmetria facciale Un certo grado è di variazione tra i due lati è")
 un nuovo guardiano del genoma • Gene di 105")
 La sindrome di Cowden è ereditata in")
 del")
 Aspetto miopatico Dovuto a scarso tono muscolare con faccia che sembra più stretta")


 Epicanto È una plica verticale di pelle ridondante tra l’occhio ed il naso.")
 Occhi infossati Possono essere un segno di un problema generale. Il neonato apre")
 Proptosi Protrusione del globo oculare • È un segno associato alla s. di")


 del")

 Sclere blu La sclera blu è normale sino ad 1 anno di età,")
 Coloboma dell’iride Fissurazione dell’iride (tipo occhi di gatto) associato a coloboma della retina")
 Occhi a mandorla La lunghezza della fessura palpebrale è diminuita • È un")


 microftalmia/anoftalmia Occhi piccoli in modo abnorme, infossati e con palpebre strette Occhi assenti")

 Posizionate in basso Sono uno dei segni più aspecifici e spesso fonte di")
 Varianti del lobo Lobo fissurato sindrome di Beckwith. Wiedemann, con macroglossia, visceromegalia, ipoglicemia")
 Polidattilia post-assiale E’ presente un dito in più sul lato ulnare della mano.")
 Polidattilia pre-assiale Duplicazione completa o parziale di un pollice normale. Si può vedere")
 Sindattilia Fusione tra le dita che può essere ossea o coinvolgere solo la")
 Clinodattilia Deflessione mediale o laterale di una o più dita. In genere mediale")
 aracnodattilia Dita più lunghe e sottili Sindrome di Marfan FBN 1 Sindrome di")
 camptodattilia Dita in posizione flessa ad una o più articolazioni. E’ spesso coinvolto")
 brachidattilia Dita più corte. L’accorciamento è in genere falangeo e si associa a")
 Dita gonfie Dita più piene, segno che può confondere Coffin-Lowry che è un")
 Dita sovrapposte Evidente sovrapposizione con la camptodattilia Trisomia 13 e trisomia 18 Beals")
 Ectrodattilia Mancanza delle dita centrali che si estende al metacarpo Ectrodattilia, displasia ectodermica")
 Pollici larghi Il pollice presenta un aspetto allargato Sindrome di Rubinstein-Taybi con microcefalia")


- Slides: 98
Semeiotica genetica Vincenzo Nigro Dipartimento di Patologia Generale Seconda Università degli Studi di Napoli Telethon Institute of Genetics and Medicine (TIGEM)
ACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTC CGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGC GACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTA GCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCG CACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCT CTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGAT ATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGAACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGC TCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGC GCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGG CTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCG ATATAGCTCGCGACACAGA TATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCAC ACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATAT ATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGA GACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACA CCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATA TAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGT AGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGA CGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAG CGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGAC GTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAG ACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCC CTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCG CTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGATAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCT AGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCT AGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTC GCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACC GCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACC GCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTC GAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGA AACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGA AACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGAC ACACACAGATATTATAGCTCGCGACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAG ACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATA GCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACA CGTGCTAGCTCCTCTCGAGACGTTATAGCTCGCGACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTA GCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGAC ACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCG AGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGC ACACCGCTCGAGATAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCT CGATATAGCTCGCGACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGACGTAGGGC TCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCT GAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACAC GTGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTC ACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACAC AGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCT CTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGA
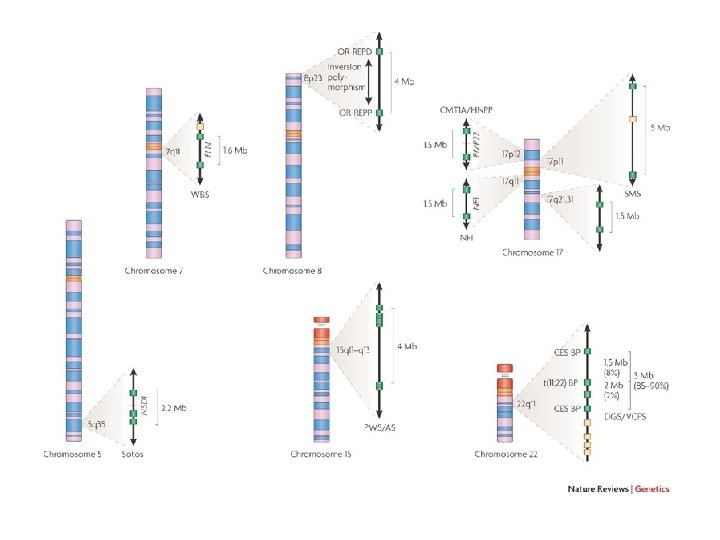
ACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTC CGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGC GACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTA GCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCG CACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCT CTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGAT ATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGAACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGC TCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGC GCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGG CTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCG ATATAGCTCGCGACACAGA TATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCAC ACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATAT ATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGA GACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACA CCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATA TAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGT AGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGA CGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAG CGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACTATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCGAGACGTAGGGCTCTCGATATA GCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGC TAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCG ACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAG CTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCT AGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACA GCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACA GCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGAC GTAGGGCTCTCGATATAGCTCGCGACA CACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTC CTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACA GATATATAGCGGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGATAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCT CCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCT CCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGA CCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCT CGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGTAGGGCTCTCGATATAGCT CGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTC CGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTCCTCT CGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATA TATAGCGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAG ATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACAC CGCTCGAGACCTTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACAC AGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATAT AGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTTATAGCTCGCGACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTG AAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACACGTGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGA Copy Number Variation 10% of the human genome could vary in copy number 1 2
duplicazioni segmentali • il genoma umano contiene complessivamente il 13, 7% di segmenti duplicati con almeno il 90% di identità di sequenza • il 5, 2% del genoma contiene segmenti duplicati lunghi tra 1 e 10 kb, mentre il 4, 5% tra 10 kb e 20 kb • i cromosomi più colpiti sono l’Y (50, 4%) ed il 22 (11, 9%), ma anche il 7, 9, 10, 15, 16, 17 e X • le duplicazioni segmentali possono essere intracromosomiche o intercromosomiche • con tre localizzazioni differenti: – pericentromeriche (47 Mb, dupliconi originati da altri cromosomi) – subtelomeriche (ciascuna solo 50 -100 kb, orientate) – interstiziali (solo nella specie umana sono disseminate ad una distanza media di 3 Mb)
Malattie autosomiche dominanti Come fanno le delezioni in uno solo dei due alleli a costituire un carattere dominante? 1. il livello dimezzato di prodotto genico è insufficiente a mantenere il fenotipo 2. il difetto eterozigote diviene omozigote a livello delle cellule dei tessuti periferici (LOH) 3. un solo allele è espresso per imprinting dell’altro
principali sindromi da delezione
Sindrome di Di. George
Di. George/velocardiofacciale La sindrome di Di. George del 22 q 11. 2 è la più frequente sindrome da microdelezione, con un incidenza di 1 su 4000— 5000 nati La delezione comprende 3 Mb ed almeno 30 geni
• Migrating neural crest cells make a contribution to the embryonic structures affected in Di. George syndrome. • The cartoon represents a human embryo at 4– 6 weeks gestation. • The migration of neural crest cells from the hindbrain to the branchial arch/pharyngeal pouch system and cardiac outflow tract is indicated by the arrows. • Examples of malformations associated with perturbation of this process are listed and these overlap substantially with those seen in 22 q 11 DS AAA, arch arteries; PDA, persistent ductus arteriosus; IAA, interrupted aortic arch.
Di. George • È caratterizzata da – Anomalie cardiache – T-cell deficit – palatoschisi – anomalie facciali – Ipocalcemia Mutazioni puntiformi del gene TBX 1 possono portare a questi 5 tratti fenotipici, ma non alle difficoltà nell’apprendimento che è invece frequente nella sindrome da delezione
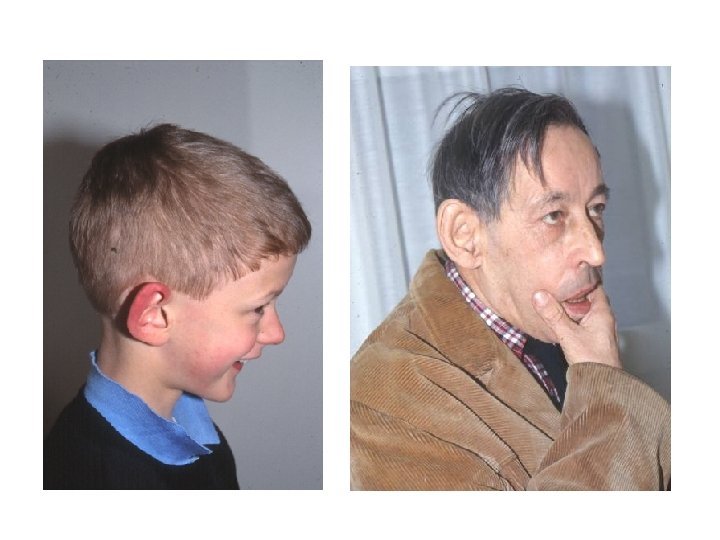
Williams-Beuren • prevalenza alla nascita 1/75001/20. 000, ma può non essere diagnosticata
Williams una delezione tipica
Williams genetica • delezione “de novo” • trasmissione autosomica dominante • delezione di 1. 6 MB da 21 geni contigui in eterozigosi a 7 q 11. 23 – gene dell’elastina – LIM kinase 1 (LIMK 1) – CLIP-115 che lega i microtubuli – Fattori di trascrizione GTF 2 IRD 1 – effetto posizionale su altri geni circostanti la delezione
Williams FISH delezione 7 q 11. 23 nrilevabile mediante FISH ma non cariotipo
Williams comportamento • lieve o medio ritardo mentale (IQ tra 41 e 80) • scarsa capacità di concentrazione • ritardo nell’apprendimento del linguaggio e poi esagerata loquacità • personalità amichevole e affettuosa • danno facilmente confidenza anche a sconosciuti • ansietà, spesso preoccupati per il benessere altrui • ipersensibilità ai suoni • memoria visiva e uditiva spesso fuori dal comune • ricordano persone, luoghi e motivi musicali • predisposizione ad imparare le lingue e la musica
Williams aspetto e segni • Faccia da elfo • Occhi blu (77%) con pattern stellato dell’iride (74%) ma questo vale per i nordeuropei, strabismo (40%) • Naso con la punta bulbosa • bocca larga e guance piene • microdontia e micrognazia • Statura 10 cm in meno del normale • ipercalcemia • stenosi periferica delle arterie polmonari • stenosi aortica sopravalvolare
http: //www. wsf. org/family/photoalbum/wsfphoto. htm Williams foto
Williams foto
Imprinting
Imprinting • Nelle cellule germinali primordiali l’imprinting viene cancellato del tutto e il DNA è demetilato • Successivamente nella linea germinale maschile si determina un pattern di imprinting che in alcuni loci è complementare a quello della linea germinale femminile • I cromosomi su cui avviene l’imprinting (7, 11, 15) manterranno questo pattern e lo riprodurranno ad ogni mitosi • Si potranno sempre distinguere l’espressione genica del cromosoma materno e paterno
Disomia uniparentale • Due copie dello stesso cromosoma sono ereditate dallo stesso genitore • Spesso questo avviene attraverso un fenomeno transitorio di trisomia, seguito dalla perdita del cromosoma singolo e mantenimento del cromosoma doppio
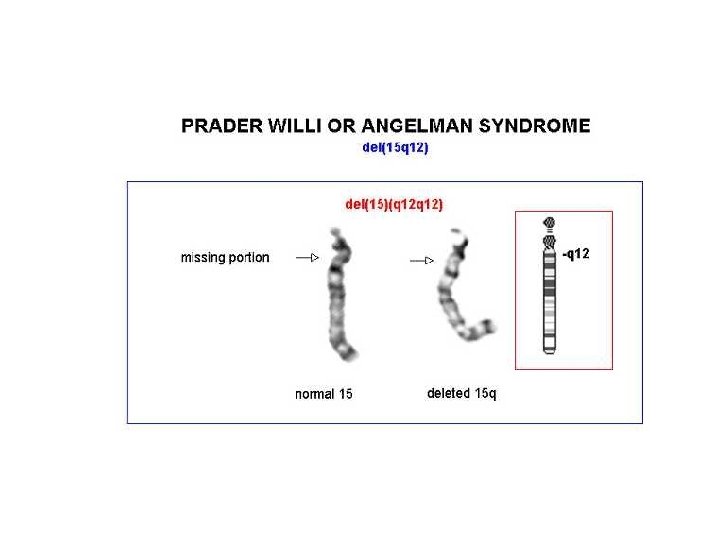
Angelman • 70% dei casi delezione della regione cromosomica 15 q 11 -q 13, che è soggetta al fenomeno dell'imprinting del cromosoma paterno • Il gene materno (l'unico espresso) può essere alterato con 4 meccanismi noti: – – delezione disomia uniparentale paterna difetti nell'imprinting mutazioni a carico del gene UBE 3 A (ubiquitin ligasi) • La diagnosi è clinica e il difetto genetico non si identifica nel 20% dei casi
Angelman • "happy puppet syndrome" si può identificare in Cucciolo (Dopey) "addormentato", il più giovane dei nani che non ha mai imparato a parlare • ritardo mentale con assenza del linguaggio, difficoltà nell'equilibrio, eccessivo buon umore
Angelman • L'incidenza è 1/20. 000 nati • crisi epilettiche e comunque alterazioni dell'EEG e microcefalia relativa
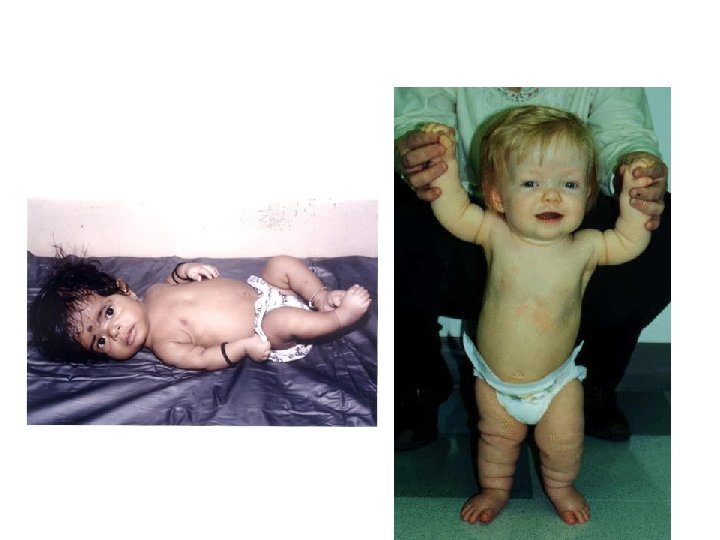
Prader-Willi • • • iperfagia>obesità eccessiva assunzione di liquidi reazioni abnormi ai sedativi acromicria, criptorchidismo insensibilità al dolore, lesioni cutanee sbalzi di umore
Prader-Willi 1/15. 000
Malattie genetiche da mutazione in 1 allele Le mutazioni monoalleliche possono causare disordini a trasmissione dominante o recessiva legata all’X negli uomini • • • Se la malattia a trasmissione dominante è grave in età fertile e pertanto limita o annulla la capacità riproduttiva (bassa fitness), le mutazioni monoalleliche sono nuove e spesso distribuite in modo casuale Se la malattia dominante non è grave in età fertile e non limita in alcun modo la capacità riproduttiva (normale fitness), le mutazioni monoalleliche sono ereditate da un genitore e spesso si tramandano da molte generazioni Se la malattia è recessiva legata all’X ed è letale ha una vita media di tre generazioni, perché le donne trasmettono gli alleli mutati in eterozigosi e gli uomini li eliminano
eredità autosomica dominante a penetranza completa (malattia che non modifica la fitness)
mutazioni puntiformi missenso • Le mutazioni missenso sono quelle in cui il cambiamento determina nel prodotto proteico la sostituzione di un aminoacido con un aminoacido differente • Sebbene queste alterazioni generalmente non provochino conseguenze nella funzionalità della proteina (polimorfismi o varianti) , ci sono casi in cui anche una minima alterazione può avere conseguenze gravi
1) plagiocefalia = asimmetria del cranio Frontale o posteriore (posturale? ) • Può essere isolata o sindromica • craniosinostosi • Analisi del cariotipo specie se i genitori sono normali • Esaminare le dita delle mani e dei piedi e cercare pollici ed alluci larghi • Guardare eventuali colorazioni della pelle (acanthosis nigricans) CRANIO
2) Cresta metopica = prominenza al centro della fronte fino alla trigonocefalia • È comunemente non patologica • Craniosinostosi della sutura metopica • Esposizione al valproato sodico • Analisi del cariotipo specie se i genitori sono normali • Esaminare le dita delle mani e dei piedi e cercare pollici ed alluci larghi • Possibile delezione crom. 11 q o 9 p CRANIO
acrocefalosindattilia sindrome di Apert • 1: 65. 000 alla nascita • craniosinostosi, volta cranica a forma conica • ipertensione endocranica • ritardo mentale • ipoplasia della parte centrale della faccia • sindattilia delle dita delle mani e dei piedi • sordità e atrofia ottica
acrocefalosindattilia sindrome di Apert • tutti i pazienti hanno la stessa mutazione Apert (Cys 755 Gly) del gene human fibroblast growth factor receptor 2 (FGFR 2) • la mutazione è in eterozigosi • de novo • cromosoma 10 q 26 • la sindrome è allelica con Crouzon e Pfeiffer
sindrome di Pfeiffer • alcuni pazienti hanno la mutazione Pfeiffer (Cys 342 Arg) del gene human fibroblast growth factor receptor 2 (FGFR 2) • altri la mutazione Pro 252 Arg in FGFR 1 • la mutazione è in eterozigosi • de novo • cromosoma 10 q 26 • la sindrome è allelica con Crouzon e Apert
disostosi cranio facciale sindrome di Crouzon • alcuni pazienti hanno la mutazione (Cys 342 Tyr) del gene human fibroblast growth factor receptor 2 (FGFR 2) • la mutazione è in eterozigosi • de novo • cromosoma 10 q 26 • la sindrome è allelica con Pfeiffer e Apert con alcune mutazioni in comune
3) Macrocefalia • È comunemente non patologica, specie se condivisa in famiglia • Può essere un segno di ipocondroplasia • Esaminare le dita delle mani e dei piedi e cercare polidattilia (Greig) • osservare la pelle • X-fragile, Cowden, Neurofibromatosi CRANIO
acondroplasia • • nanismo dismorfico (1: 35. 000) arti corti e testa sproporzionatamente più grossa fronte prominente e naso appiattito altezza media 130 cm nei maschi 125 cm nelle femmine • La mutazione è in eterozigosi • Gly 380 Arg nel recettore 3 del "fibroblast growth factor" (FGFR 3) a 4 p 16. 3 • autosomico dominante a penetranza completa
acondroplasia • La mutazione conferisce una funzione aumentata al recettore dell'FGF (allele ipermorfo) che è una tirosinchinasi di membrana • In risposta all'FGF il recettore dimerizza e si fosforila trasducendo un segnale con la funzione di rallentare la proliferazione dei condrociti e quindi la crescita ossea • Topi senza il gene FGF 3 R hanno ossa lunghe e vertebre allungate
ipocondroplasia • L'ipocondroplasia ha caratteristiche simili all'acondroplasia, ma di gravità minore con un coinvolgimento craniofacciale inferiore. L'altezza può risultare ai limiti della norma e la malattia viene spesso non diagnosticata. • L'ipocondroplasia è meno omogenea: circa il 70% dei casi è dovuto alla sostituzione N 540 K del gene FGFR 3, mentre non si conosce la mutazione nel restante 30%.
Circa il 2% della popolazione ha un IQ<70 (ritardo mentale) il 15 -20% di tutti I ritardi mentali sono attribuibili a geni del cromosoma X Il ritardo mentale legato al cromosoma X (XLMR) è geneticamente eterogeneo con 202 loci responsabili di forme che si sovrappongono clinicamente 46 geni sono stati a tutt’oggi identificati il locus che contribuisce alla frazione maggiore causa la sindrome di Martin. Bell, oggi nota come sindrome dell’X fragile
X fragile ritardo mentale: IQ tra 20 e 70 • deficit di memoria a breve termine di informazioni complesse • ritardo nel linguaggio • ridotte abilità visuo-spaziali • ipersensibilità agli stimoli • iperattività con deficit di attenzione • comportamento autistico • Macrocefalia con fronte, mento e orecchie sporgenti • Macroorchidismo (<30 ml) dopo la pubertà • Anomalie connettivali: prolasso della mitrale, lassità articolare, piede piatto • Disfunzioni ipotalamiche?
Nel 1969 Lubs osservò una costrizione (marker X) sul braccio lungo del cromosoma X in quattro maschi affetti e tre carriers obbligate della stessa famiglia
Il sito fragile a Xq 27. 3 rottura o costrizione dei cromosomi in metafase che insorge quando le cellule sono esposte ad una perturbazione della replicazione del DNA siti fragili sono su tutti i cromosomi e prendono il nome della banda cromosomica, es fra(X)(q 27. 3) la nomenclatura HUGO chiama questo sito FRAXA, cioè il primo sito fragile identificato sul cromosoma X
Segregazione, paradosso di Sherman Il 20% dei maschi che portano l’allele mutato sono normali (NTM) Il 30% delle carrier presenta ritardo mentale 1 perché è affetta? I 2 1 II perché non è affetto? 4 3 1 III 1 IV 2 3 4 5
Fragile X syndrome
CRANIO 4) Microcefalia • È comunemente autosomica recessiva, ma può essere dovuta a sostanze teratogene • Può essere un segno di un un disturbo generale della crescita • Esaminare sindattilia (Smith-Lemli. Opitz o Filippi), ipoplasia pollici e pigmentazione cutanea (Fanconi), orecchie prominenti (Richarson-Kirk)
CRANIO 5) Varianti della linea di attaccatura dei capelli • Ciuffo ribelle (cowlick) • Escludere craniosinostosi • Attaccatura frontale alta (cariotipo delezione 8 q, ATRX) • Attaccatura bassa e widow’s peak (displasia cranio-facciale) • attaccatura posteriore bassa (Noonan o Turner) dovuta ed edema nucale prenatale
Monosomia X (45, X 0) Turner 1: 2. 500 • Solo l’ 1% delle gravidanze giunge a termine • Errore nella spermatogenesi nell’ 80% dei casi e non correla con l’età dei genitori • Caratteristiche principali: – mancato sviluppo ovarico con amenorrea primaria e sterilità – fenotipo molto variabile – linfedema con rigonfiamento delle mani e dei piedi – pterigio del collo
Monosomia X (45, X 0) Turner 1: 2. 500 – mandibola più piccola (micrognazia) – torace largo con aumento degli spazi intercostali – attaccatura bassa delle orecchie – bassa statura – quarto metacarpo corto – cardiopatia, ipertensione e anomalie renali – sia l’intelligenza sia l’attesa di vita sono normali
6) Ipertelorismo = occhi più distanziati maggiore sviluppo relativo delle ali dello sfenoide, per cui le orbite sono più distanziate tra loro: in tal caso la radice del naso è allargata • Da valutare se ci sia telecanto, cioè lo spostamento laterale del canto interno • Può essere associato al “Widow’s peak” e può essere familiare e benigno • Osservare la madre se presenta ipertelorismo più lieve (Opitz, ATRX) • Associato alla bassa statura e a brachidattilia (Aarskog) OCCHI
mutazioni eterozigoti di PAX 3 Waardenburg
mutazioni eterozigoti di PAX 3 Waardenburg • sordità (o deficit uditivo di vario livello) bilaterale, • modifiche nella pigmentazione, sia dei capelli (albinismo parziale, in genere piebaldismo) che della pelle, • anomalie nello sviluppo dei tessuti derivati dalla cresta neurale • lateralizzazione del canto mediale • diverso colore degli occhi (eterocromia), di solito uno marrone e l'altro blu
7) Ipotelorismo = occhi più vicini Ridotta distanza interpupillare, spesso associato a sindromi cromosomiche • Da valutare se ci sia telecanto che riduce l’impressione dei ipotelorismo • Può essere arrivare sino alla ciclopia, nel caso di oloprosencefalia, con mancato sviluppo del tratto olfattorio • Il 40% della oloprosencefalia è dovuto alla trisomia del cromosoma 13 • Escludere la sindrome di Kallmann (+ipogonadismo e anosmia) e le delezioni a Xp OCCHI
trisomia 13 Patau http: //www. livingwithtrisomy 13. org • (1/12. 000 -20. 000 nati) • 90% dei casi nondisgiunzione materna • Giunge a termine solo il 2. 5% dei concepimenti • Di questi il 33% muore nel primo mese, il 50% entro 2 mesi • Peso sotto la norma, difficoltà suzione • Oloprosencefalia, microcefalia • Cecità e sordità • Occhi che possono fondersi • Labiopalatoschisi 80% • epilessia • Malformazioni cardiache • sinclinodattilia • piedi a calcagno prominente
8) sinofri = sopracciglia che si incontrano sulla linea mediana È spesso associato ad ipotelorismo • Nel bambino è un segno della sindrome di “Cornelia de Lange”, con problemi gastrointestinali, ritardo, ecc • Valutare ev mucopolisaccaridosi SOPRACCIGLIA
10) Micrognazia = mandibola più MANDIBOLA piccola Si vede meglio di profilo a bocca aperta • Sequenza di Pierre-Robin, con palatoschisi e spostamento posteriore della lingua per mancato sviluppo mandibolare • Sindrome di Stickler, associata a miopia con distacco di retina, artropatia (dovuta a mutazioni AD di geni del collageno) • Aneuplodia anche nel caso di mosaicismo
Monosomia X (45, X 0) Turner 1: 2. 500 – mandibola più piccola (micrognazia) – torace largo con aumento degli spazi intercostali – attaccatura bassa delle orecchie – bassa statura – quarto metacarpo corto – cardiopatia, ipertensione e anomalie renali – sia l’intelligenza sia l’attesa di vita sono normali
11) Asimmetria facciale Un certo grado è di variazione tra i due lati è comune e nella vita in utero può essere dovuta ad oligoidramnios • Eventuale craniosinostosi (Pfeiffer) • Può essere dovuta ad emiiperplasia isolata e i bambini non hanno altro • Valutare le orecchie e le dita • esiste la sindrome asymmetric crying facies (ACFS) in cui l’asimmetria si vede al pianto • Può essere una manifestazione della sindrome di Proteus, associata a abnorme crescita ossea, cutanea e muscolare, a volta con mutazioni di PTEN FACCIA
PTEN (phosphatase and tensin homolog) un nuovo guardiano del genoma • Gene di 105 kb a 10 q 23 • PTEN è una fosfatasi doppia (dual-specificity phosphatase) agendo – sia sugli aminoacidi, serina, treonina e tirosina – sia sui lipidi, rimuovendo il fosfato D 3 dell’inositolo (fosfatidilinositolo-3 -fosfato fosfatasi) • ha funzioni di oncosoppressore – inibendo la via del segnale basata su AKT / PKB – mantenendo la stabilità cromosomica • PTEN è perduta per mutazioni, delezioni o o silenziamento del promotore in molte neoplasie primitive e metastatiche (prostata, mammella, ovaio, esofago, pancreas, ecc)
un solo allele mutato di PTEN (eterozigosi) La sindrome di Cowden è ereditata in modalità autosomica dominante I pazienti presentano più amartomi e lesioni in vari organi e tessuti Cancro della mammella e/o carcinoma follicolare della tiroide Macrocefalia ed anomalie del cervelletto papule sul dorso di mani e piedi papillomatosi della mucosa orale
sindrome di Pfeiffer • alcuni pazienti hanno la mutazione Pfeiffer (Cys 342 Arg) del gene human fibroblast growth factor receptor 2 (FGFR 2) • altri la mutazione Pro 252 Arg in FGFR 1 • la mutazione è in eterozigosi • de novo • cromosoma 10 q 26 • la sindrome è allelica con Crouzon e Apert
11) Aspetto miopatico Dovuto a scarso tono muscolare con faccia che sembra più stretta e bocca aperta • Può essere distrofia miotonica, con storia di polidramnios, in tal caso osservare la madre con attenzione e valutare il fenomeno miotonico al pollice • Può essere una patologia muscolare mitocondriale • Sindrome di Moebius, con paralisi facciale ed incapacità al movimento laterale degli occhi, per deficit dei nervi VI e VII FACCIA
anticipazione nella distrofia miotonica
distrofia miotonica DM 1 • fenomeno “miotonico”, difficoltà al rilasciamento muscolare dopo una contrazione • ipotonia al volto, non debolezza importante • cataratta precoce • alterazioni ritmo cardiaco • disfunzione tiroidea • trasmissione autosomica dominante (1/8000) • forma congenita con grave ipotonia neonatale
1) Epicanto È una plica verticale di pelle ridondante tra l’occhio ed il naso. Si origina sotto l’occhio e si estende in alto verso la palpebra. Spesso il canto mediale viene oscurato • Può essere normale se è depresso il ponte nasale e scompare entro i due anni • Si osserva nela s. di Down ed in altre aneuploidie • Si può osservare nella s. di Williams, nella s. di Ehlers-Danlos e di Stickler associata a micrognazia, miopia con distacco di retina, artropatia (dovuta a mutazioni AD di geni del collageno) OCCHIO
2) Occhi infossati Possono essere un segno di un problema generale. Il neonato apre gli occhi nei primi 2 gg, se non c’è fotofobia o microlftamia. Valutare di profilo l’assenza di curvatura del globo oculare • Nella s. di Freeman-Sheldon si associa a camptodattilia • Nella s. di Lowe (X-linked) al ritardo mentale, tubulopatia ed aminoaciduria: mutazioni del gene OCRL che regola la formazione dell’actina nelle giunzioni della lente e dei tubuli prossimali • Delezioni di 1 p 36, con obesità, ispessimento dell’elice con asimmetria OCCHIO
3) Proptosi Protrusione del globo oculare • È un segno associato alla s. di Crouzon se non c’è alterazione delle dita, altrimenti altre craniosinostosi come Pfeiffer (alluci e pollici larghi) ed Apert (con sindattilia) • Ehlers-Danlos, con alterazioni cutanee, iperlassità articolare, dovuta a mutazioni del gene lisil idrossilasi • Unilaterale, neurofibromatosi tipo I OCCHIO
acrocefalosindattilia sindrome di Apert • 1: 65. 000 alla nascita • craniosinostosi, volta cranica a forma conica • ipertensione endocranica • ritardo mentale • ipoplasia della parte centrale della faccia • sindattilia delle dita delle mani e dei piedi • sordità e atrofia ottica
acrocefalosindattilia sindrome di Apert • tutti i pazienti hanno la stessa mutazione Apert (Cys 755 Gly) del gene human fibroblast growth factor receptor 2 (FGFR 2) • la mutazione è in eterozigosi • de novo • cromosoma 10 q 26 • la sindrome è allelica con Crouzon e Pfeiffer
sindrome di Pfeiffer • alcuni pazienti hanno la mutazione Pfeiffer (Cys 342 Arg) del gene human fibroblast growth factor receptor 2 (FGFR 2) • altri la mutazione Pro 252 Arg in FGFR 1 • la mutazione è in eterozigosi • de novo • cromosoma 10 q 26 • la sindrome è allelica con Crouzon e Apert
disostosi cranio facciale sindrome di Crouzon • alcuni pazienti hanno la mutazione (Cys 342 Tyr) del gene human fibroblast growth factor receptor 2 (FGFR 2) • la mutazione è in eterozigosi • de novo • cromosoma 10 q 26 • la sindrome è allelica con Pfeiffer e Apert con alcune mutazioni in comune
4) Sclere blu La sclera blu è normale sino ad 1 anno di età, dopo può essere importante • È un segno associato alla osteogenesi imperfetta, con fratture e ritardo della dentizione • Ehlers-Danlos, con alterazioni cutanee, iperlassità articolare, dovuta a mutazioni del gene lisil idrossilasi • sindrome di Marfan, dovuta a mutazioni della fibrillina FBNI con aracnodattilia, apparenza magra • Bambino molto piccolo alla nascita: Silver Russell OCCHIO
5) Coloboma dell’iride Fissurazione dell’iride (tipo occhi di gatto) associato a coloboma della retina da cui deriva embriologicamente • CHARGE: coloboma, heart, atresia choana, retardation, genital, ear malfomation con mutazioni di CDH 7 (chromodomain helicase DNA binding protein 7) • Wolf-Hirschhorn: ipertelorismo, con delezione 4 p • Neurofibromatosi I OCCHIO
6) Occhi a mandorla La lunghezza della fessura palpebrale è diminuita • È un segno associato alla sindome di Prader-Willi, con ipotonia neonatale, acromicrìa ed obesità dopo i 6 anni • Delezioni di 1 p 36, con obesità, ispessimento dell’elice con asimmetria OCCHIO
Wolf-Hirschhorn delezione a 4 p 16. 3
Wolf-Hirschhorn • • • Scarso accrescimento Ritardo mentale, ipotonia Labbro leporino • Conformazione ad elmo di guerriero greco
7) microftalmia/anoftalmia Occhi piccoli in modo abnorme, infossati e con palpebre strette Occhi assenti • TORCH, alcol e warfarin • Nella s. di Lowe (X-linked) al ritardo mentale, tubulopatia ed aminoaciduria: mutazioni del gene OCRL che regola la formazione dell’actina nelle giunzioni della lente e dei tubuli prossimali • Sindrome oculo dento digitale, con ipotelorismo, sindattilia cutanea OCCHIO
ORECCHIO elice antelice trago lobo L’architettura dell’orecchio esterno è soggetto a grande variabilità. Molte varianti non hanno alcun significato patologico
1) Posizionate in basso Sono uno dei segni più aspecifici e spesso fonte di confusione. La radice dell’elice passa sotto una linea orizzontale immaginaria che unisce i canti laterali • Sindrome di Noonan con bassa statura, dismorfismo facciale, malformazioni cardiache, deformità del torace e pterigio: mutazioni di PTPN 11 crom. 12 q 24. 1 • Nella s. di Rubinstein-Taybi, delez 16 p, con ritardo mentale pollici larghi, orecchie malformate Orecchio
2) Varianti del lobo Lobo fissurato sindrome di Beckwith. Wiedemann, con macroglossia, visceromegalia, ipoglicemia neonatale e delezioni a 11 p 15 Lobo assente Ehlers-Danlos, con alterazioni cutanee, iperlassità articolare, dovuta a mutazioni del gene lisil idrossilasi Lobo molto più grande sindrome di Kabuki, con obesità, ipotonia, problemi cardiaci, s. di Costello Lobo rivolto verso l’alto Mowat-Wilson, con Hirschsprung, epilessia, microcefalia, malattia cardiaca congenita da delezioni del gene Zinc finger E-box binding homeobox 2 Orecchio
1) Polidattilia post-assiale E’ presente un dito in più sul lato ulnare della mano. Può essere piccolo e sottile ed essere costituito da una proiezione di tessuto tra le articolazioni metacarpofalangee e interfalangee prossimali Patau (trisomia 13) con palatoschisi e/o soffi cardiaci Bardet-Biedl (BBS) con obesità al primo anno di vita, ipogenitalismo e poi distrofia retinica dopo i 10 anni Mano
2) Polidattilia pre-assiale Duplicazione completa o parziale di un pollice normale. Si può vedere un secondo pollice adiacente. Se la duplicazione è limitata alla falange ditale, si può vedre un’unica larga unghia. Esaminare anche i piedi Effetti di sostane teratogene e diabete materno Sindrome di Robinow con ipertelorismo, palatoschisi Sindrome di Towness-Brocks (TBS) con malformazioni anali, alterazioni auricolari, insufficienza renale e mutazioni del gene SALL 1 (Sal-like 1, zinc finger) Mano
3) Sindattilia Fusione tra le dita che può essere ossea o coinvolgere solo la pelle tra le dita Sindrome di Apert (FGFR 2) Sindattilia cutanea può essere dovuta a triploidia o mosaicismo con triploidia Sindrome di Timothy con allungamento del tratto QT per mutazioni del gene CACNA 1 C Mano
4) Clinodattilia Deflessione mediale o laterale di una o più dita. In genere mediale del V dito. Osservare le giunzioni interfalangee che curvano l’una verso l’altra Sindrome di Down nel 60% dei casi Duplicazioni e delezioni cromosomiche varie Sindrome di Silver-Russel con basso peso e lunghezza, ma normale circonferenza cranica (disomia 7 o ipometilazione a 11 p 15) Mano
5) aracnodattilia Dita più lunghe e sottili Sindrome di Marfan FBN 1 Sindrome di Beals FBN 2, con camptodattilia e contratture, appiattimento dell’elice Mano
6) camptodattilia Dita in posizione flessa ad una o più articolazioni. E’ spesso coinvolto il V dito e l’articolazione interfalangea Sindrome di Beals FBN 2, con camptodattilia e contratture, appiattimento dell’elice Sindrome miastenica materna Mano
7) brachidattilia Dita più corte. L’accorciamento è in genere falangeo e si associa a clinodattilia Sostanze teratogene come alcool e fenitoina Sindrome di de Lange Sindrome di Robinow con ipertelorismo, spostamento dell’ombelico verso lo sterno e mutazioni del gene ROR 2 Sindrome di Aarskog Brachidattilia familiare Mano
8) Dita gonfie Dita più piene, segno che può confondere Coffin-Lowry che è un ritardo mentale legato all’X con ipotonia, ipertelorismo e pienezza delle labbra e mutazioni del gene RSK 2. Le dita non sono doloranti Mano
9) Dita sovrapposte Evidente sovrapposizione con la camptodattilia Trisomia 13 e trisomia 18 Beals sindrome con mutazioni FBN 2 Mano
10) Ectrodattilia Mancanza delle dita centrali che si estende al metacarpo Ectrodattilia, displasia ectodermica e palatoschisi EEC sindrome con pelle secca, ipercheratosi, ipodontia, iride blu e fotofobia ed eredità autosomica dominante da mutazioni di p 63 Mano
11) Pollici larghi Il pollice presenta un aspetto allargato Sindrome di Rubinstein-Taybi con microcefalia alluce largo fessure palbebrali che vanno verso il basso Pfeiffer Apert Brachidattilia autosomica dominante Mano
Progeria Hutchinson-Gilford • invecchiamento precoce • bassa statura, pelle rugosa • calvizie, assenza di tessuto adiposo • aterosclerosi ed infarto
Progeria Hutchinson-Gilford • nuova mutazione in eterozigosi del gene lamina A • la mutazione è in eterozigosi • de novo • cromosoma 1 q 23 • La mutazione non cambia l'aminoacido glicina G 608 G, ma introduce un sito donor di splicing GGT che fa perdere 50 aminoacidi alla proteina • sperimentazione con inibitori di farnesil-trasferasi