Roles of RNA m RNA messenger r RNA
 • r. RNA (ribosomal) • t. RNA")


 i < j < k <")

 or = i j-1 j i k")


 Quantity Fitting Function Parameters Groundstate")
 This figure shows the frozen pairs")








 uses 15 nuclear plus 3 mitochondrial proteins")

 Gene uses 12 mitochondrial proteins")
 53 complete Mammalian mitochondrial genomes Complete set of r.")










- Slides: 35
Roles of RNA • m. RNA (messenger) • r. RNA (ribosomal) • t. RNA (transfer) • other ribonucleoproteins (e. g. spliceosome, signal recognition particle, ribonuclease P) • viral genomes • artificial ribozymes
Typical transfer RNA structure
Thermodynamics parameters are measured on real molecules. Helix formation = hydrogen bonds + stacking G C U A C U Multi-branched loop Bulges Internal loops } DG = -1. 2 kcal/mol U loop DG = + 4. 5 kcal/mol C Entropic penalty for loop formation. } DG = -2. 1 kcal/mol Sum up contributions of helices and loops over the whole structure. Hairpin loop
Pairs i-j and k-l are compatible if (a) i < j < k < l , or (b) i < k < l < j. (c) is called a pseudoknot: i < k < j < l. Usually not counted as secondary structure. (b) (a) (c) k k i k l l l j i i j j Bracket notation is used to represent structure: a: ((((. . )))). . ((((. . )))) b: ((. ((((. . )))). )) Basic problem: Want an algorithm that considers every allowed secondary structure for a given sequence and finds the lowest energy state.
Simplest case: find structure which maximizes number of base pairs. Let = -1 if bases can pair and + if not. Ignore loop contributions. E(i, j) = energy of min energy structure for chain segment from i to j. We want E(1, N). or = i j-1 j i k j Algorithms that work by recursion relations like this are called dynamic programming. The algorithm is O(N 3) although the number of structures increases exponentially with N. Also need to do backtracking to work out the minimum energy structure: Set B(i, j) = k if j is paired with k, or 0 if unpaired.
Partition Function Algorithm (for simplest energy rules) or = i j-1 j i k where Real Energy Rules : Need to consider many special cases. What type of loop are you closing? Algorithm is more complex but still is O(N 3). j
Equilibrium probability that base i is paired with j 1 i j Equilibrium probability that base i is unpaired Example of pairing probabilities taken from Vienna package web-site N
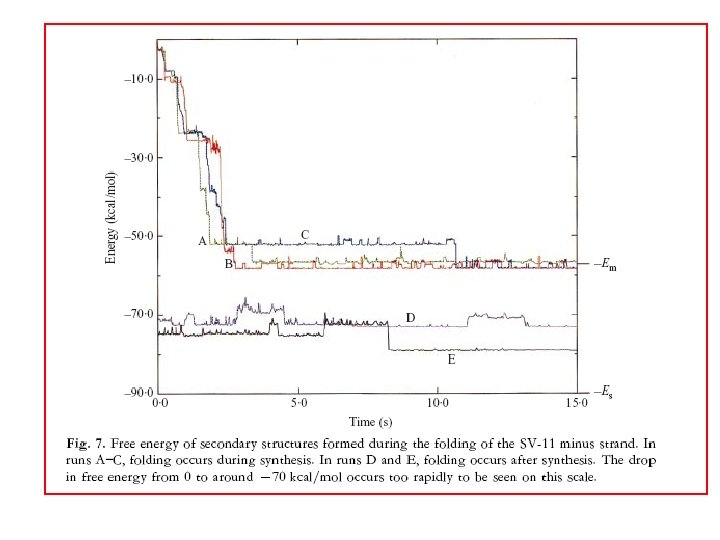
Is folding kinetics important? RNA folding kinetics involves reorganisation of secondary structure i A C B D iii I ii B E B C D F H D G Native structures may not be global minimum free energy states. Morgan & Higgs (1996) J. Chem. Phys.
Energy Landscapes in RNA Folding Morgan & Higgs (1998) Quantity Fitting Function Parameters Groundstate energy C 1 = 2. 9 ( 0. 2) = -0. 368 ( 0. 001) Total number of states C 2 = -5. 6 ( 0. 4) = 0. 533 ( 0. 001) Number of groundstates C 3 = 1. 75 ( 0. 2) = 0. 068 ( 0. 001) Groundstates are degenerate in this model because energies are integers. Generate many random groundstates. How far apart are these groundstates? How high are the barriers between groundstates?
We found Frozen pairs (present in every groundstate) This figure shows the frozen pairs only. The molecule is divided into independent unfrozen loops. Define Neff as the length of the longest loop. Two groundstates for the same sequence
Minimum Free Energy Prediction Deterministic. Always gets MFE structure for a given set of energy rules. If MFE structure is not the same as biological structure, this could be because (i) energy rules are inaccurate or insufficient (ii) kinetics is important and molecule is trapped in metastable state. Monte Carlo simulations of folding kinetics. Store a current structure. Estimate rates of removal of existing helices and rates of addition of other compatible helices. Choose one helix to be added or removed with probability proportional to its rate. Repeat this many times. Can simulate structure formation from an unfolded state.
Q is a bacteriophage RNA virus with approx 4000 nucleotides Viral RNA has complex secondary structure. The replicase gene codes for the replicase protein. This is an RNA-dependent RNA polymerase. Synthesizes complementary strand. Viral replication needs two steps: plus to minus to plus.
In vitro RNA evolution in the Q system c Begin with Replicase + nucleotides + viral RNA c Replicase + nucleotides only Transfer small quantity to each successive tube c c sequence RNA after many transfers
Barrier heights between alternative groundstates Observation: Mean barrier height between groundstates scales as <h> ~ Neff 0. 5 Neff ~ 0. 3 N Therefore barriers become significant for large enough sequences.
An example where kinetics is important to control biological function: the 5’ region of the MS 2 phage. 3500 130 Maturation protein
Time to formation of the 5’ structure influences expression of the maturation protein more than the stability of this structure. Simulations compare with experiments on mutant sequences.
RNA in comparison to Proteins Both have well defined 3 d structures RNA folding problem is easier because secondary structure separates from tertiary structure more easily - But it is still a complex problem. RNA model has real parameters therefore you can say something about real molecules. RNA folding algorithm is simple enough to be able to do statistical physics. (cf. 27 -mer lattice protein models).
Part of sequence alignment of Mitochondrial Small Sub-Unit r. RNA Full gene is length ~950 11 Primate species with mouse as outgroup
Murphy et al. Nature (2001) uses 15 nuclear plus 3 mitochondrial proteins
Afrotheria / Laurasiatheria Striking examples of convergent evolution
Cao et al. (2000) Gene uses 12 mitochondrial proteins
RNA pairs model (GR 7) 53 complete Mammalian mitochondrial genomes Complete set of r. RNAs + t. RNAs from = 973 pairs. 100 86 100 97 100 Jow et al. (2002)
MCMC searches the rugged landscape in tree space using the Metropolis algorithm. Obtains a set of possible trees weighted according to their likelihood. 1. Rate parameter changes = continuous 2. Branch length changes = continuous E A 3. Topology changes = discrete D 2 E C A A E Nearest-neighbour interchange D C B D 1 4 B Long-range move E B C C D 3 B A
Models of Sequence Evolution rij is the rate of substitution from state i to state j States label bases A, C, G & T Pij(t) = probability of being in state j at time t given that ancestor was in state i at time 0. i t j
The HKY model describes rate of evolution of single sites to from The frequencies of the four bases are k is the transition-transversion rate parameter * means minus the sum of elements on the row
Compensatory Substitutions Two sides of the acceptor stem from a t. RNA are shown. Due to structure conservation alignment is possible in widely different species. Bacillus subtilis Escherichia coli Saccharomyces cerevisiae Drosophila melanogaster Homo sapiens 1234567 ((((((( 7654321 ))))))) GGCUCGG GCCCGGA GCGGAUU GCCGAAA CCGAGCC UCCGGGC AAUUCGC UUUCGGC
Model 7 A is a General Reversible 7 -state Model 7 frequencies pi + 21 rate parameters ij - 2 constraints = 26 free parameters
Probability of remaining in same state Pii SSU r. RNA sequences from Eubacteria
Probability Pij of changes from CG to other pairs SSU r. RNA from Eubacteria
What is going on? AU GU UA fast GC fast slow UG CG Selection against GU and UG is weaker than against mismatches. Double transitions are faster than double transversions. Double transitions are faster than single transitions to GU and UG states. This is explained by theory of compensatory substitutions.
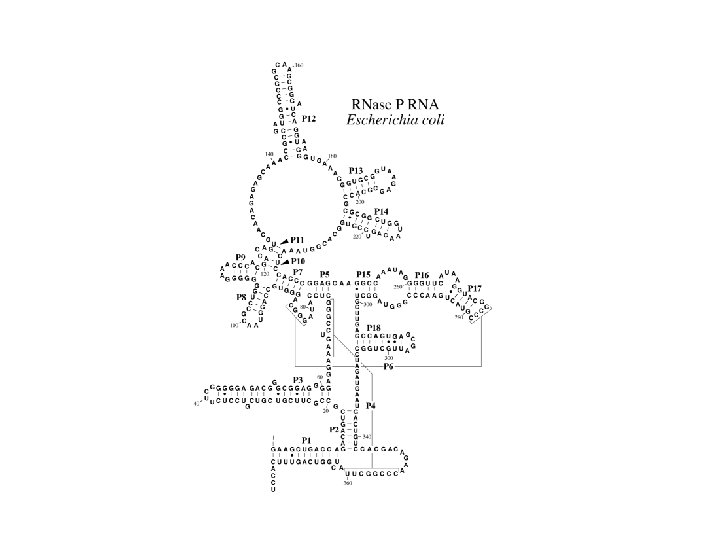
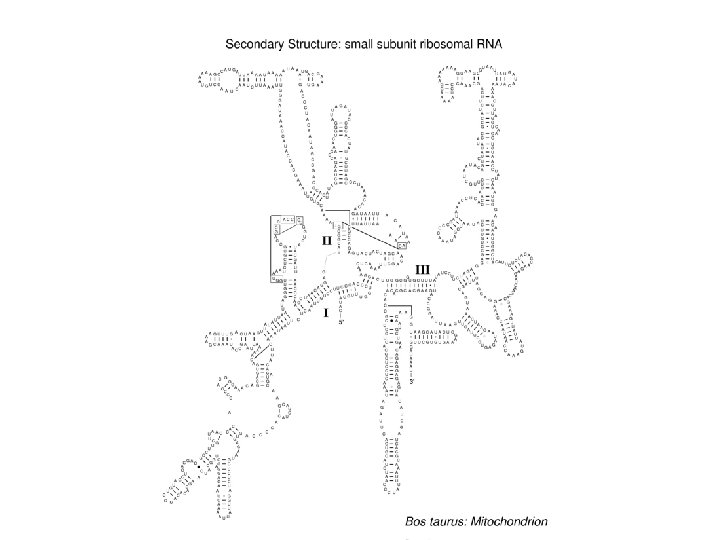
Analysis of RNA sequence databases t. RNA mitoch. t. RNA general t. RNA archaea Rnase P SSU r. RNA G+C average G+C helical regions 0. 339 0. 448 0. 532 0. 681 0. 636 0. 829 0. 594 0. 730 0. 545 0. 674 Frequencies 0. 266 0. 121 0. 257 0. 233 0. 046 0. 030 0. 046 0. 372 0. 260 0. 128 0. 142 0. 043 0. 025 0. 030 0. 473 0. 320 0. 057 0. 077 0. 031 0. 020 0. 022 0. 385 0. 296 0. 117 0. 104 0. 050 0. 022 0. 026 0. 352 0. 298 0. 122 0. 173 0. 020 0. 021 0. 014 Number of sequences 884 754 64 84 455 Number of pairs 21 21 21 80 296 GC CG AU UA GU UG MM Selection for thermodynamically stable structures Higgs (2000) Quart. Rev. Biophysics
Analysis of RNA Substitution Rates t. RNA mitoch. t. RNA general t. RNA archaea Rnase P SSU r. RNA 0. 67 0. 84 0. 86 0. 77 2. 44 3. 32 2. 32 0. 49 0. 83 1. 46 1. 24 1. 96 5. 01 0. 99 0. 45 0. 89 4. 01 1. 78 1. 85 3. 00 0. 86 0. 65 0. 60 1. 46 1. 09 1. 72 2. 84 5. 24 0. 55 0. 66 1. 40 0. 93 3. 92 4. 36 7. 84 Double transitions / Double transversions 4. 7 1. 7 2. 3 3. 1 2. 1 Double transitions / Transitions to GU or UG 1. 6 2. 0 8. 9 3. 6 2. 8 Mutabilities GC CG AU UA GU UG MM Thermodynamic properties influence Evolutionary properties