Regulatory Issues Introduction to the Regulatory Approval Process


















 are the methods by which manufacturers, holders,")










 Each person engaged in the manufacture, processing,")








 or less • Same matrix")





 processed through the")










- Slides: 56
Regulatory Issues
• • • • Introduction to the Regulatory Approval Process; Overview of the FDA Investigational New Drug Application (IND); Summary of regulations and guidelines Introduction of c. GMP's/principles of validation Introduction to QA/QC principles Good Laboratory Practice (GLP) compliance Pre-clinical testing for biotechnology products; safety and toxicology Clinical stages, design of clinical trials and protocols, evaluation of clinical data Regulatory Filings: Biological License Application (BLA) Pre-approval inspections Team Biologics International regulatory status for biotechnology products; WHO, Japan, and the EC/CPMP application International Conference on Harmonization (ICH) update Regulatory considerations for gene therapy and transgenic products
FDA Structure / Organization Food and Drug Administration Office of Combination Products Center for Veterinary Devices National Center for Toxicological Research Center for Biologics Evaluation and Research Center for Food Safety and Applied Nutrition Center for Devices and Radiological Health Center for Drug Evaluation and Research
CDRH Offices Office of Device Evaluation Office of Surveillance & Biometrics Office of Compliance Office of In-Vitro Diagnostic Devices & Safety Center for Devices and Radiological Health Office of Science & Technology Office of Health & Industry Programs
FDA's Three Key Development Roles: • "Gatekeeper" to the marketplace -- the new drug approval process • "Cop on the beat" or "Enforcer" -- ensuring quality compliance via inspection and enforcement actions (e. g. criminal charges) • "Sentinel" of Safety Concerns - during development and post-approval
FDA regulation of medical products • Among the products that FDA regulates are three categories of diagnostic, preventative, or therapeutic products: – Drugs – Biologics – Medical devices 6
The Approval Gate … • Preliminary Considerations -- Determining the Regulatory Status of the product – Is it a "drug", "device" or "biologic"? • Drug: – described in USP (United States Pharmacopeia) or – intended (via labeling) » to affect the body of man or other animals » to be used in the diagnosis, cure, mitigation, treatment or prevention of disease in man or other animals
The Approval Gate … • Regulatory Status of the product - con'd… – Is it a "drug", "device" or "biologic"? • Device: defined as involving: "instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or "similar or related article including any component, part or accessory. " – – – in USP/NF (the National Formulary) or intended to be used in diagnosis … cure, mitigation, treatment or prevention of disease or other conditions intended to affect the body of man
The Approval Gate … • Regulatory Status of the product - con'd… – Is it a "drug", "device" or "biologic"? • Thus -- device definition capture products that resemble drugs if they do not achieve their result via being metabolized in the body or via chemical action within or on the body -- regulated by FDA Center for Devices & Radiological Health (CDRH) – Examples of "drug-like" devices: » Ultrasound contrast media » Contact lens solutions
The Approval Gate … • Regulatory Status of the product - con'd… – Is it a "drug", "device" or "biologic"? • Biologic -– – – • Generally, if derived from human or animal tissue; used to be regulated by FDA Center for Biologics (CBER) using approval standards similar to CDER therapeutic biotech products going to CDER » vaccines – remain behind NOTE: "true" biotech products usually are biologics
The Approval Gate … • Regulatory Status of the product - con'd… – Is it a "drug", "device" or "biologic"? – OR BOTH? ? – "Combination" or "hybrid" products -- • are regulated per their "primary mode of action" - • but this may be difficult to discern -- get clarification very early as will impact FDA Center you deal with • can request in writing -- under FDAMA § 416, FDA can't later change its mind w/o your consent or public health reasons exist
The Approval Gate … • Regulatory Status of the product - con'd… – What type of submission is needed to get FDA approval or clearance? – Drugs: • • Full New Drug Application (NDA) 505(b)(2) NDA or "Paper NDA" Abbreviated New Drug Application The OTC Drug route -- Abreva (Avanir/SKB) – NDA – OTC Review monograph change
The Approval Gate … • Regulatory Status of the product - con'd… – What type of submission is needed to get FDA approval or clearance? – Devices: • Premarket Approval Application (PMA) -- clinical studies will be needed • Premarket Notification under § 510 k -- clinical studies MAY be needed (or wanted)
The Approval Gate … • Regulatory Status of the product - con'd… – What type of submission is needed to get FDA approval or clearance? – Biologics • Biologic License Application (BLA) • no generic versions now possible – may change …
The Approval Gate … • Regulatory Status of the product - con'd… – What quantity and quality of data will be demanded by FDA to show safety & effectiveness? – Will vary -- FDA has extensive discretion here – Key task -- try to get clarity as soon as possible in the process -- Ways to do so: • • Pre-IND meeting -- encouraged by FDA prior to start of human clinicals End of Phase 2 Meeting - also encouraged -- here's where you want to "lock" them in
Overview of Typical Pharmaceutical Product Development IP Marketing Research Pre. Clinical Work Cost: 1 F I L I N G Preclinical to Phase II - Approximately 1 -7 million Marketing Plan Product Launch A P P R O V A L Clinical Studies Phase III - 2 - 8 million V A L I D A T I O N Registration Validation Batches - Product Costs and Labor X 3 to 5 batches Commercial Production Total Costs = 10 -25 million USD Production Start Up Costs based on Contract or Facility Total Time = 4. 5 - 7. 5 years Time: 2, 3 Preclinical to Completed Clinicals - 3 -5 years FDA Approval - 13. 5 months 3 Validation and Production Launch - 6 -18 months In 2000 Dollars - Estimates by the National Cancer Institute for all new pharmaceutical. Estimate does not consider R&D costs that are not associated with the development of the drug in question. Most drug companies use a system of cost estimates that includes the valuation of money if it had been invested andthe cost of drug development not approved by the FDA. Most studies conclude that the rate of commercialization success to be 1: 5000. How Much does it cost to develop a new drug - James Love Consumer Project on Technology http: //www. cptech. org April 2, 2000 2 Drug Approval Overregulation, MR Ward - CATO Regulation - http: //www. cato. org/pubs/regulation/reg 15 n 4 e. htm 3 New York Times - November 8 1995 1 16
Welcome to the Jungle Effective Ineffective GO FAIL Animal Ineffective Inferior FAIL Toxic GO Chemistry FAIL Degradants & Impurities on ati Phase IV lid Ineffective Effective Failure is Unlikely GO FAIL GO Sell Product Va Passes ID & Description Definition Nominal Batch SS Grows Bugs ULATE FAIL Launch PA Micro REFORM GO GO FDA STUDIES Phase III Death of Product Antimicrobial & Aseptic Effective Ineffective GO Fill Sales & Warehouse Pipeline SS Toxicology FAIL Efficacy FAIL RESET ALL PARAMETERS GO Max. Energy Batch PA Safe Pa Rese ram t ete rs Phase II FAIL Launch Ad Campaign GO GO ff Proof of Concept FAIL SS Unstable PA FAIL Scale Up Production Min. Energy Batch n. O Stability Phase I Commercial Production Pre Approval Inspection Sig GO a ul m r o te k F ua ea val Tw e-E R Registration Validation Approval s Stable Clinical Trials Filing Pa Rese ram t ete r Pre Clinical Work Reformulation Egg START OVER Clinical Report Validation Report Quarantine Product Formula Improvement Geriatric or Pediatric Stability Testing Drug Interaction 17 Define LT & Side Effects
And the next step… • You’ve got the device or drug okayed—now you have to manufacture it…
GMPs • Current good manufacturing practices (GMPs) are the methods by which manufacturers, holders, and transporters of drugs, biologics, or devices assure that every product that they make, hold, or transport is, and continues to be until it is used, safe and effective. • Failure to comply with GMPs (and for devices, failure to comply with the quality system regulations) makes a product “adulterated” and its distribution or sale illegal. 19
The Early Beginnings • 1900 s house-calls • Home remedies, ointments and “miracle elixirs” • Entertainment and music • No regulations until 1902 Fig. 1. Animation of ancient medicine show
Public Involvement • 1905 - The Jungle by Upton Sinclair • Exposure of unsanitary conditions in meat packing plants • Public awareness and involvement Fig. 2. The Jungle by Upton Sinclair • Pure Food and Drug Act • False labeling became illegal Fig. 3. 1906 Meat processing plant
What is GMP? • Good Manufacturing Practice is a set of regulations, codes, and guidelines for the manufacture of drug substances and drug products, medical devices, in vivo and in vitro diagnostic products, and foods. Fig. 4 GMP handbooks for every industry
Good Manufacturing Practices Worldwide Enforcement • Good Manufacturing Practices are enforced in the United States by the FDA • In the United Kingdom by the Medicines and Healthcare Products Regulatory Agency • GMPs are enforced in Australia by the Therapeutically Goods Administration • In India by the Ministry of Health, multinational and/or foreign enterprises • Many underdeveloped countries lack GMPs
1941 Initiation of GMP • Sulfathiaziole tablets contaminated with phenobarbital • 1941 - 300 people died/injured • FDA to enforce and revise manufacturing and quality control requirements • 1941 - GMP is born Fig. 5 1906 Certificate of Purity signed by doctor
1962 Kefauver-Harris Drug Amendments • Thalidomide tragedy • Thousands of children born with birth defects due to adverse drug reactions of morning sickness pill taken by mothers • Strengthen FDA’s regulations regarding experimentation on humans and proposed new way how drugs are approved and regulated • “Proof of efficacy” law
1976 Medical Device Amendments • 1972 and 1973 -Pacemaker failures reported • 1975 - hearing-Dalkon Shield intrauterine device caused thousands of injuries • Class I, II and III medical devices – based on degree of control necessary to be safe and effective Fig. 7 President Gerald Ford signs the Medical Device Amendments
1980 Infant Formula Act • 1978 - major manufacturer of infant formula reformulated two of its soy products • 1979 - Infants diagnosed with hypochloremic metabolic alkalosis • Greater regulatory control over the formulation and production of infant formula • Modification of industry’s and FDA’s recall procedures Fig. 8 Parody on Infant Formula Act
Part 211 –Selected c. GMP For Finished Pharmaceuticals • • • Subpart E-Control of Components and Drug Product Containers and Closures 211. 80 General requirements. 211. 82 Receipt and storage of untested components, drug product containers, and closures. 211. 84 Testing and approval or rejection of components, drug product containers, and closures. 211. 86 Use of approved components, drug product containers, and closures. Subpart F-Production and Process Controls 211. 100 Written procedures; deviations. 211. 101 Charge-in of components. 211. 103 Calculation of yield. 211. 105 Equipment identification. • . . . • • • • Subpart A-General Provisions Subpart B-Organization and Personnel 211. 22 Responsibilities of quality control unit. 211. 25 Personnel Qualifications. 211. 28 Personnel responsibilities. Subpart C-Buildings and Facilities 211. 46 Ventilation, air filtration, air heating and cooling. 211. 58 Maintenance Subpart D-Equipment 211. 63 Equipment design, size, and location. 211. 65 Equipment construction. 211. 67 Equipment cleaning and maintenance. 211. 68 Automatic, mechanical, and electronic equipment. 211. 72 Filters. • •
§ 211. 25 Personnel qualifications • (a) Each person engaged in the manufacture, processing, packing, or holding of a drug product shall have education, training, and experience, or any combination thereof, to enable that person to perform the assigned functions. Training shall be in the particular operations that the employee performs and in current good manufacturing practice (including the current good manufacturing practice regulations in this chapter and written procedures required by these regulations) as they relate to the employee's functions. Training in current good manufacturing practice shall be conducted by qualified individuals on a continuing basis and with sufficient frequency to assure that employees remain familiar with CGMP requirements applicable to them. • (b) Each person responsible for supervising the manufacture, processing, packing, or holding of a drug product shall have the education, training, and experience, or any combination thereof, to perform assigned functions in such a manner as to provide assurance that the drug product has the safety, identity, strength, quality, and purity that it purports or is represented to possess. • (c) There shall be an adequate number of qualified personnel to perform and supervise the manufacture, processing, packing, or holding of each drug product.
Quality Assurance vs. Quality Control Quality Assurance An overall management plan to guarantee the integrity of data (The “system”) Quality Control A series of analytical measurements used to assess the quality of the analytical data (The “tools”)
True Value vs. Measured Value True Value The known, accepted value of a quantifiable property Measured Value The result of an individual’s measurement of a quantifiable property
Accuracy vs. Precision Accuracy Precision How well a measurement agrees with an accepted value How well a series of measurements agree with each other
Systematic vs. Random Errors Systematic Error Avoidable error due to controllable variables in a measurement. Random Errors Unavoidable errors that are always present in any measurement. Impossible to eliminate
Quality Control Measures • • • Standards and Calibration Blanks Recovery Studies Precision and Accuracy Studies Method Detection Limits State Laws
Standards and Calibration • • • Prepared vs. Purchased Standard Signals: Peak Area, Beer’s Law Calibration Curves Continuing Calibration Checks Internal Standards Performance Testing
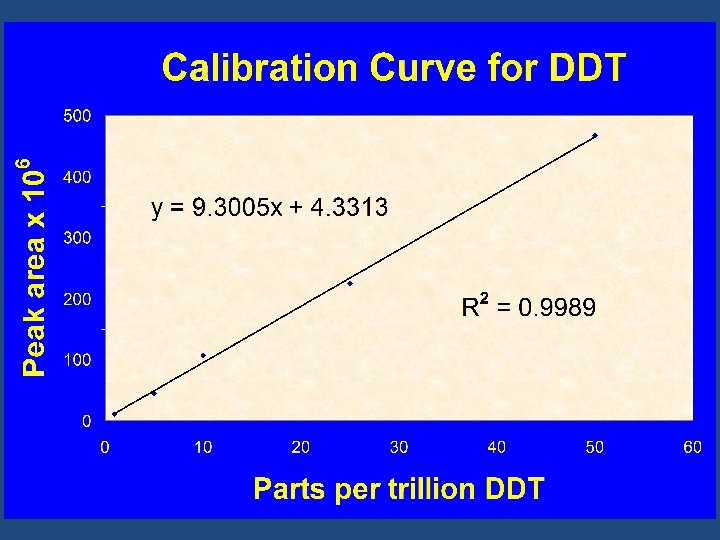
Calibration Curves Graphical representation of the relationship between: • The concentration of the analyte and • The analytical signal
Continuing Calibration Verification • Many methods don’t require that daily calibration curves are prepared • A “calibration verification” is analyzed with each batch of samples
Sample Batch • 10 - 20 samples (method defined) or less • Same matrix • Same sample prep and analysis • Contains a full set of QC samples
Internal Standards • A compound chemically similar to the analyte • Not expected to be present in the sample • Cannot interfere in the analysis • Added to the calibration standards and to the samples in identical amounts
Internal Standards • Refines the calibration process • Analytical signals for calibration standards are compared to those for internal standards • Eliminates differences in random and systematic errors between samples and standards
Performance Testing Blind samples submitted to laboratories ? Labs must periodically analyze with acceptable results in order to maintain accreditation ? ?
Blanks, Blanks • Laboratory Reagent Blanks • Instrument Blanks • Field Reagent Blanks • Trip Blanks
Laboratory Reagent Blanks • Contains every reagent used in the analysis • Is subjected to all analytical procedures • Must give signal below detection limit • Most methods require one with every batch
Instrument Blank • A clean sample (e. g. , distilled water) processed through the instrumental steps of the measurement process; used to determine instrument contamination
Field Reagent Blanks • Prepared in the lab, taken to the field • Opened at the sampling site, exposed to sampling equipment, returned to the lab.
Trip Blanks • Prepared in the lab, taken to the field • Not opened • Returned to the lab • Not always required in EPA methods
Recovery Studies • Matrix Spikes • Laboratory Control Samples • Surrogates
Matrix Spikes • Sample spiked with a known amount of analyte • Subjected to all sample prep and analytical procedures • Determines the effect of the matrix on analyte recovery • Normally one per batch
Laboratory Control Sample • Analyte spiked into reagent water • Subjected to all sample prep and analytical procedures
Precision and Accuracy • Required for initial certification and annually thereafter • A series of four laboratory control samples • Must meet accuracy (recovery) and precision (standard deviation) requirements, often in method
Method Detection Limit “The minimum concentration of a substance that can be measured and reported with 99% confidence that the analyte concentration is greater than zero”
Method Detection Limit • MDLs are determined according to 40 CFR, part 136, Appendix B • Seven replicate laboratory control samples, analyzed for precision
Method Detection Limit • Must be performed initially for certification • Must meet criteria specified in method • Must be performed with change in instrumentation or test method • Annually with ELCP (Environmental Laboratory Certification Program)
State Laws • Each state has laws governing laboratories and their personnel.