RBC and BLEEDING DISORDERS RBC and Bleeding Disorders

RBC and BLEEDING DISORDERS

RBC and Bleeding Disorders • NORMAL – Anatomy, histology – Development – Physiology • ANEMIAS – Blood loss: acute, chronic – Hemolytic – Diminished erythropoesis • • POLYCYTHEMIA BLEEDING DISORDERS
 Hemoglobin")
TABLE 13 -2 -- Adult Reference Ranges for Red Blood Cells Measurement (units) Hemoglobin (gm/d. L) Hematocrit (%) Red cell count (10 /µL) 6 Men 13. 6– 17. 2 Women 12. 0– 15. 0 39– 49 33– 43 4. 3– 5. 9 3. 5– 5. 0 Reticulocyte count (%) 0. 5– 1. 5 Mean cell volume (µm ) MCV 82– 96 Mean corpuscular hemoglobin (pg) MCH 27– 33 Mean corpuscular hemoglobin concentration (gm/d. L) MCHC 33– 37 3 RBC distribution width * 11. 5– 14. 5

WHERE is MARROW? • Yolk Sac: very early embryo • Liver, Spleen: NEWBORN • BONE – CHILDHOOD: AXIAL SKELETON & APPENDICULAR SKELETON BOTH HAVE RED (active) MARROW – ADULT: AXIAL SKELETON RED MARROW, APPENDICULAR SKELETON YELLOW MARROW

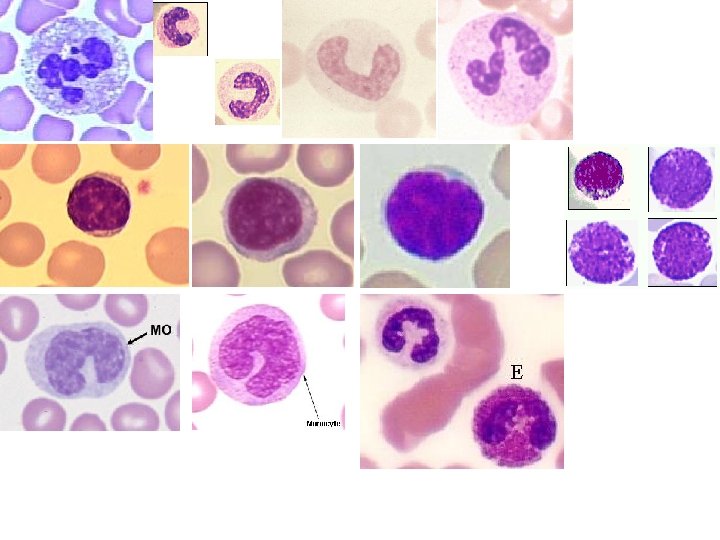
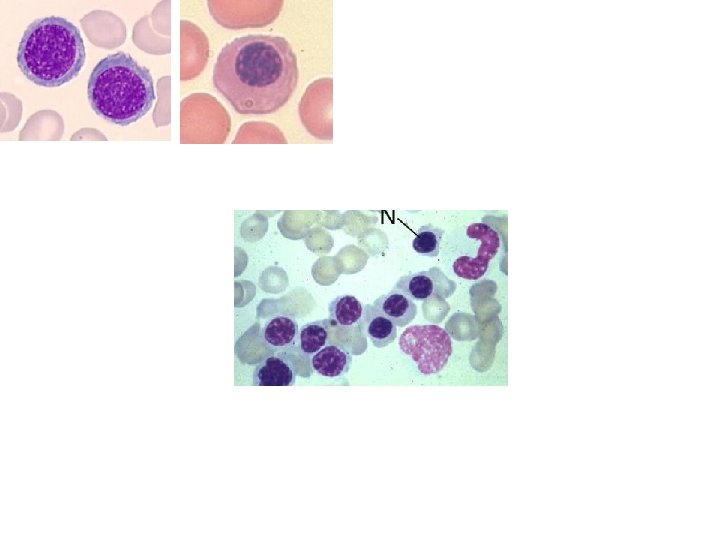
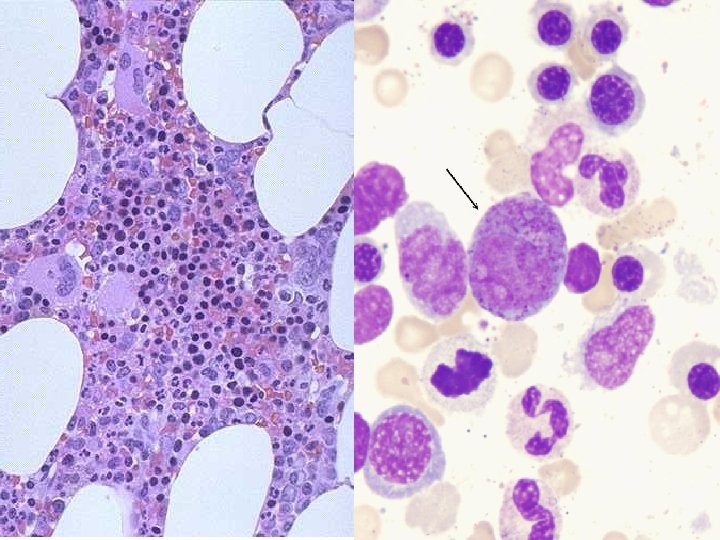
MARROW FEATURES • • CELLULARITY 50% MEGAKARYOCYTES at least 1 -2/hpf M: E RATIO 3: 1 MYELOID MATURATION 1/3 bands or more ERYTHROID MATURATION nucleus/cytoplasm LYMPHS, PLASMA CELLS small percentage STORAGE IRON, i. e. , HEMOSIDERIN present “FOREIGN CELLS”

MARROW “DIFFERENTIATION”
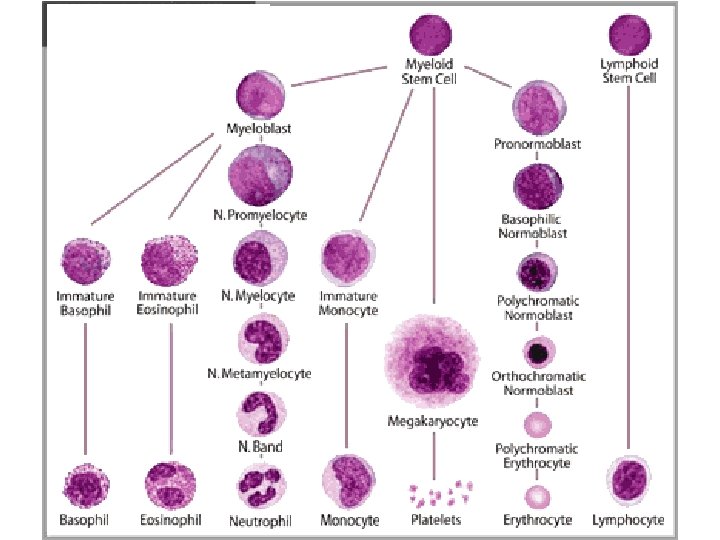
 • DE-creased")
ANEMIAS* • BLOOD LOSS – ACUTE – CHRONIC • IN-creased destruction (HEMOLYTIC) • DE-creased production * A good definition would be a decrease in OXYGEN CARRYING CAPACITY, rather than just a decrease in red blood cells, because you need to have enough blood cells THAT FUNCTION, and not just enough blood cells.

Features of ALL anemias • Pallor, where? • Tiredness • Weakness • Dyspnea, why? • Palpitations • Heart Failure (high output), why?

Blood Loss Acute: trauma Chronic: lesions of gastrointestinal tract, gynecologic disturbances. The features of chronic blood loss anemia are the same as iron deficiency anemia, and is defined as a situation in which the production cannot keep up with the loss.

HEMOLYTIC • HEREDITARY – MEMBRANE disorders: e. g. , spherocytosis – ENZYME disorders: e. g. , G 6 PD deficciency – HGB disorders (hemoglobinopathies) • ACQUIRED – MEMBRANE disorders (PNH) – ANTIBODY MEDIATED, transfusion or autoantibodies – MECHANICAL TRAUMA – INFECTIONS – DRUGS, TOXINS – HYPERSPLENISM

IMPAIRED PRODUCTION • Disturbance of proliferation and differentiation of stem cells: aplastic anemias, pure RBC aplasia, renal failure • Disturbance of proliferation and maturation of erythroblasts • Defective DNA synthesis: (Megaloblastic) • Defective heme synthesis: (Fe) • Deficient globin synthesis: (Thalassemias)

MODIFIERS • MCV, microcytosis, macrocytosis • MCHC, hypochromic • RDW, anisocytosis
,")
HEMOLYTIC ANEMIAS • Life span LESS than 120 days • Marrow hyperplasia (M: E), EPO+ • Increased catabolic products, e. g. , bilirubin, serum HGB, hemosiderin, haptoglobin-HGB
 • EXTRA-vascular (spleen)")
HEMOLYSIS • INTRA-vascular (vessels) • EXTRA-vascular (spleen)

M: E Ratio normally 3: 1

HEREDITARY SPHEROCYTOSIS Genetic defects affecting ankyrin, spectrin, usually autosomal dominant Children, adults Anemia, hemolysis, jaundice, splenomegaly, gallstones (what kind? )
 Deficiency • A and Mediterranean are most significant")
Glucose-6 -Phosphate Dehydrogenase (G 6 PD) Deficiency • A and Mediterranean are most significant types

FEATURES of G 6 PD Defic. • Genetic: Recessive, X-linked • Can be triggered by foods (fava beans), oxidant substances drugs (primaquine, chloroquine), or infections • HGB can precipitate as HEINZ bodies • Acute intravascular hemolysis can occur: – Hemoglobinuria – Hemoglobinemia – Anemia

Sickle Cell Disease • • Classic hemoglobinopathy Normal HGB is α 2 β 2: β-chain defects (Val->Glu) Reduced hemoglobin “sickles” in homozygous 8% of American blacks are heterozygous
 Vaso-occlusive")
Clinical features of HGB-S disease • • Severe anemia Jaundice PAIN (pain CRISIS) Vaso-occlusive disease: EVEREWHERE, but clinically significant bone, spleen (autosplenectomy) • Infections: Pneumococcus, Hem. Influ. , Salmonella osteomyelitis

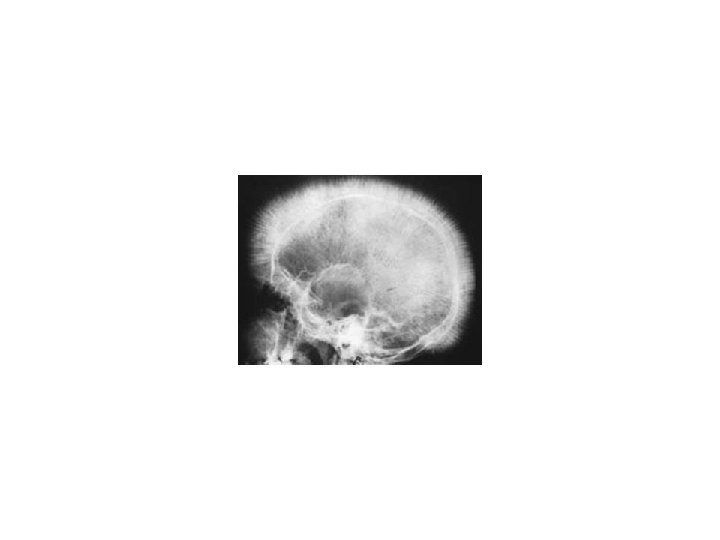
THALASSEMIAS • A WIDE VARIETY of diseases involving GLOBIN synthesis, COMPLEX genetics • Alpha or beta chains deficient synthesis involved • Often termed MAJOR or MINOR, depending on severity, silent carriers and “traits” are seen • HEMOLYSIS is uniformly a feature, and microcytic anemia, i. e, LOW MCV (just like iron deficiency anemia has a low MCV) • A “crew cut” skull x-ray appearance may be seen in severe erythroid hyperplasia.

Hemoglobin H Disease • Deletion of THREE alpha chain genes • HGB-H is primarilly Asian H HIGH • HGB- has a affinity for oxygen • HGB-H is unstable and therefore has classical hemolytic behavior

HYDROPS FETALIS • FOUR alpha chain genes are deleted, so this is the MOST SEVERE form of thalassemia • Many/most never make it to term • Children born will have a SEVERE hemolytic anemia as in the erythroblastosis fetalis of Rh disease: – Pallor (as in all anemias), jaundice, kernicterus – Edema (hence the name “hydrops”) – Massive hepatosplenomegaly (hemolysis)
 Glycosylphos. Phatidyl. Inositol (lipid rafts) • ACQUIRED, NOT INHERITED like")
Paroxysmal Nocturnal Hemoglobinuria (PNH) Glycosylphos. Phatidyl. Inositol (lipid rafts) • ACQUIRED, NOT INHERITED like all the previous hemolytic anemias were • ACQUIRED mutations in phosphatidylinositol glycan A (PIGA) • Note: It is “P” and “N” only 25% of the time!

Immunohemolytic Anemia • All of these have the presence of antibodies and/or compliment present on RBC surfaces • NOT all are AUTOimmune, some are caused by drugs • Antibodies can be – WARM (Ig. G) – COLD AGGLUTININ (Ig. M) – COLD HEMOLYSIN (paroxysmal) (Ig. G)
, will NOT agglutinate at room temp –")
IMMUNOHEMOLYTIC ANEMIAS • WARM AGGLUTININS (Ig. G), will NOT agglutinate at room temp – Primary Idiopathic (most common) – Secondary (Tumors, especially leuk/lymph, drugs) • COLD AGGLUTININS: (Ig. M), WILL agglutinate at room temp – Mycoplasma pneumoniae, HIV, mononucleosis • COLD HEMOLYSINS: (Ig. G) Cold Paroxysmal Hemoglobinuria, hemo-LYSIS in body, ALSO often follows mycoplasma pneumoniae

COOMBS TEST • DIRECT: Patient’s CELLS are tested for surface Ab’s • INDIRECT: Patient’s SERUM is tested for Ab’s.

HEMOLYSIS/HEMOLYTIC ANEMIAS DUE TO RBC TRAUMA • Mechanical heart valves breaking RBC’s • MICROANGIOPATHIES: – TTP – Hemolytic Uremic Syndrome

NON-Hemolytic Anemias: i. e. , DE-creased Production • • “Megaloblastic” Anemias B 12 Deficiency (Pernicious Anemia) Folate Deficiency Iron Deficiency Anemia of Chronic Disease Aplastic Anemia “Pure” Red Cell Aplasia OTHER forms of Marrow Failure
 from macrocytes (peripheral smear, MCV>94) • Impaired DNA")
MEGALOBLASTIC ANEMIAS • Differentiating megaloblasts (marrow) from macrocytes (peripheral smear, MCV>94) • Impaired DNA synthesis • For all practical purposes, also called the anemias of B 12 and FOLATE deficiency • Often VERY hyperplastic/hypercellular marrow

Decreased intake Inadequate diet, vegetarianism Impaired absorption Intrinsic factor deficiency Pernicious anemia Gastrectomy Malabsorption states Diffuse intestinal disease, e. g. , lymphoma, systemic sclerosis Ileal resection, ileitis Competitive parasitic uptake Fish tapeworm infestation Bacterial overgrowth in blind loops and diverticula of bowel Increased requirement Pregnancy, hyperthyroidism, disseminated cancer

Vit-B 12 Physiology • Oral ingestion • Combines with INTRINSIC FACTOR in the gastric mucosa • Absorbed in the terminal ileum • DEFECTS at ANY of these sites can produce a MEGALOBLASTIC anemia
, and that")
Please remember that ALL megaloblastic anemias are also MACROCYTIC (MCV>94 or MCV~100), and that not only are the RBC’s BIG and hyperplastic/hypercellular, but so are the neutrophils, and neutrophilic precursors in the bone marrow too, and even more so, HYPERSEGMENTED!!!

PERNICIOUS ANEMIA • • MEGALOBLASTIC anemia LEUKOPENIA and HYPERSEGS JAUNDICE NEUROLOGIC posterolateral spinal tracts ACHLORHYDRIA Can’t absorb B 12 LOW serum B 12 Flunk Schilling test, i. e. , can’t absorb B 12, using a radioactive tracer

FOLATE DEFICIENCY MEGALOBLASTIC AMEMIAS • • • Decreased Intake: diet, etoh-ism, infancy Impaired Absorption: intestinal disease DRUGS: anticonvulsants, BCPs, CHEMO Increased Loss: Hemodialysis Increased Requirement: Pregnancy, infancy Impaired Usage

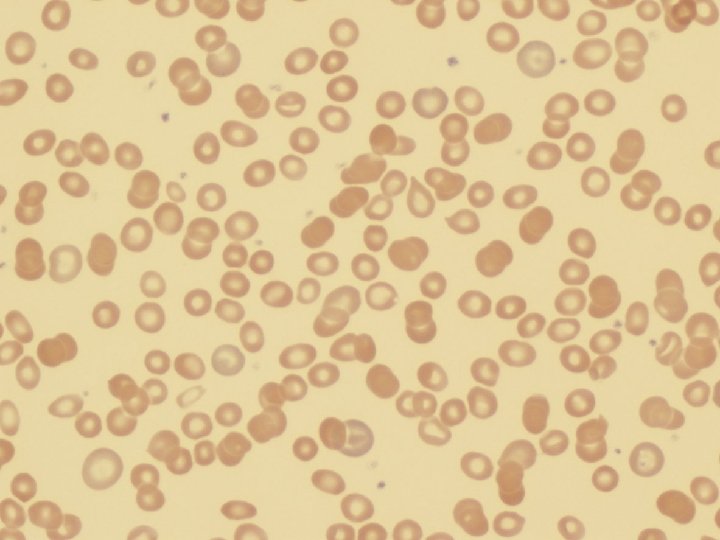
Fe Deficiency Anemia • Due to increased loss or decreased ingestion, almost always, in USA, nowadays, increased loss is the reason • Microcytic (low MCV), Hypochromic (low MCHC) • THE ONLY WAY WE CAN LOSE IRON IS BY LOSING BLOOD, because FE is recycled!
 Hemosiderin")
Fe Transferrin Ferritin (GREAT test) Hemosiderin

Clinical Fe-Defic-Anemia • Adult men: GI Blood Loss • PRE menopausal women: menorrhagia • POST menopausal women: GI Blood Loss

2 BEST lab tests: • Serum Ferritin • Prussian blue hemosiderin stain of marrow (also called an “iron” stain)
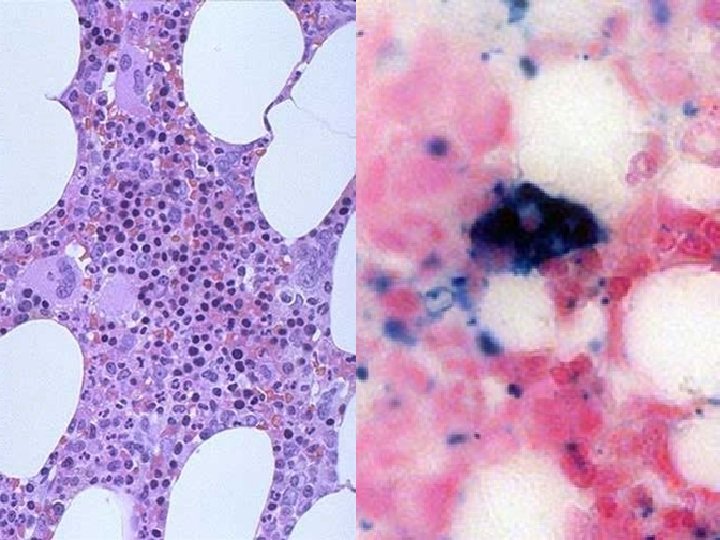

Anemia of Chronic Disease* • CHRONIC INFECTIONS • CHRONIC IMMUNE DISORDERS • NEOPLASMS • LIVER, KIDNEY failure * Please remember these patients may very much look like iron deficiency anemia, BUT, they have ABUNDANT STAINABLE HEMOSIDERIN in the marrow!

APLASTIC ANEMIAS • ALMOST ALWAYS involve platelet and WBC suppression as well • Some are idiopathic, but MOST are related to drugs, radiation • FANCONI’s ANEMIA is the only one that is inherited, and NOT acquired • Act at STEM CELL level, except for “pure” red cell aplasia

APLASTIC ANEMIAS

APLASTIC ANEMIAS • • • CHLORAMPHENICOL OTHER ANTIBIOTICS CHEMO INSECTICIDES VIRUSES – EBV – HEPATITIS – VZ

MYELOPHTHISIC ANEMIAS • Are anemias caused by metastatic tumor cells replacing the bone marrow extensively
 • Absolute – POLYCYTHEMIA VERA (Primary) (LOW")
POLYCYTHEMIA • Relative (e. g. , hemoconcentration) • Absolute – POLYCYTHEMIA VERA (Primary) (LOW EPO) – POLYCYTHEMIA (Secondary) (HIGH EPO) • HIGH ALTITUDE • EPO TUMORS • EPO “Doping” • CVAC, the trendy California bubble pods

P. VERA • A “myeloproliferative” disease • ALL cell lines are increased, not just RBCs
 • Blood vessel wall abnormalities √ • Reduced platelets")
BLEEDING DISORDERS (aka, Hemorrhagic “DIATHESES”) • Blood vessel wall abnormalities √ • Reduced platelets √ • Decreased platelet function √ • Abnormal clotting factors √ • DIC (Disseminated INTRA-vascular Coagulation), also has ↓ plats.
 (NON-thrombotic cytopenic purpuras) Infections, especially, meningococcemia, and rickettsia Drug")
VESSEL WALL ABNORMALITIES (angiopathic thrombocytopenias) (NON-thrombotic cytopenic purpuras) Infections, especially, meningococcemia, and rickettsia Drug reactions causing a leukocytoclastic vasculitis Scurvy, Ehlers-Danlos, Cushing syndrome Henoch-Schönlein purpura (mesangial Ig. A deposits too) Hereditary hemorrhagic telangiectasia (Osler–Weber– Rendu syndrome, Autosomal Dominant) • Amyloid • • •
 –")
THROMBOCYTOPENIAS • Like RBCs: – DE-creased production – IN-creased destruction – Sequestration (Hypersplenism) – Dilutional • Normal value 150 K-300 K

DE-CREASED PRODUCTION • • • APLASTIC ANEMIA ACUTE LEUKEMIAS ALCOHOL, THIAZIDES, CHEMO MEASLES, HIV MEGALOBLASTIC ANEMIAS MYELODYSPLASTIC SYNDROMES (PRELeukemias)
 POST-TRANSFUSION (NEONATAL) QUINIDINE, HEPARIN, SULFA MONO, HIV DIC,")
IN-CREASED DESTRUCTION • • AUTOIMMUNE (ITP) POST-TRANSFUSION (NEONATAL) QUINIDINE, HEPARIN, SULFA MONO, HIV DIC, “CONSUMPTIVE” TTP/HUS “MICROANGIOPATHIC”
 • Acute Immune • DRUG-induced • HIV associated")
THROMBOCYTOPENIAS • ITP (Idiopathic Thrombocytopenic Purpura) • Acute Immune • DRUG-induced • HIV associated • TTP, Hemolytic Uremic Syndrome

• • I. T. P. ADULTS AND ELDERLY ACUTE OR CHRONIC AUTO-IMMUNE ANTI-PLATELET ANTIBODIES PRESENT • INCREASED MARROW MEGAKARYOCYTES • Rx: STEROIDS
 ALSO have")
ACUTE ITP • • CHILDREN Follows a VIRAL illness (~ 2 weeks) ALSO have anti-platelet antibodies Platelets usually return to normal in a few months

DRUGS • Quinine • Quinidine • Sulfonamide antibiotics • HEPARIN

HIV • BOTH DE-creased production AND IN-creased destruction factors are present

Thrombotic Microangiopathies • BOTH are very SERIOUS CONDITIONS with a HIGH mortality: – TTP (THROMBOTIC THROMBOCYTOPENIC PURPURA) – H. U. S. (HEMOLYTIC UREMIC SYNDROME) • These can also be called “consumptive” coagulopathies, just like a DIC
: – Bernard-Soulier syndrome (Glycoprotein-1 b deficiency) –")
“QUALITATIVE” platelet disorders • Mostly congenital (genetic): – Bernard-Soulier syndrome (Glycoprotein-1 b deficiency) – Glanzmann’s thrombasthenia (Glyc. -IIB/IIIA deficiency) – Storage pool disorders, i. e. , platelets misfunction AFTER they degranulate • ACQUIRED: ASPIRIN, ASPIRIN

BLEEDING DISORDERS due to CLOTTING FACTOR DEFICIENCIES • NOT spontaneous, but following surgery or trauma • ALL factor deficiencies are possible • Factor VIII and IX both are the classic X-linked recessive hemophilias, A and B, respectively • ACQUIRED disorders often due to Vitamin-K deficiencies (II, VII, IX, X) • von Willebrand disease the most common, 1%

von Willebrand Disease • • 1% prevalence, most common bleeding disorder Spontaneous and wound bleeding Usually autosomal dominant Gazillions of variants, genetics even more complex • Prolonged BLEEDING TIME, NL platelet count • v. WF is von Willebrand Factor, which complexes with Factor VIII, it is the von Willebrand Factor which is defective in von Willebrand disease • Usually BOTH platelet and Factor. VIII-v. WF disorders are present

PTT PT/INR

• • • HEMOPHILIA A The “classic” HEMOPHILIA Factor VIII decreased Co-factor of Factor IX to activate Factor X Sex-linked recessive Hemorrhage usually NOT spontaneous Wide variety of severities • Prolonged PTT (intrinsic) only • Rx: Recombinant Factor VIII

• • • HEMOPHILIA B The “Christmas” HEMOPHILIA Factor IX decreased Sex-linked recessive Hemorrhage usually NOT spontaneous Wide variety of severities • Prolonged PTT (intrinsic) only • Rx: Recombinant Factor IX

DIC, Disseminated INTRA-vascular, Coagulation • • ENDOTHELIAL INJURY WIDESPREAD FIBRIN DEPOSITION HIGH MORTALITY ALL MAJOR ORGANS COMMONLY INVOLVED

DIC, Disseminated INTRA-vascular, Coagulation • Extremely SERIOUS condition • NOT a disease in itself but secondary to many conditions – Obstetric: MAJOR OB complications, toxemia, sepsis, abruption – Infections: Gm-, meningococcemia, RMSF, fungi, Malaria – Many neoplasms, acute promyelocytic leukemia – Massive tissue injury: trauma, burns, surgery • “Consumptive” coagulopathy
 • PT INR (extrinsic) • Platelet count, aggregation")
Common Coagulation TESTS • PTT (intrinsic) • PT INR (extrinsic) • Platelet count, aggregation • Bleeding Time, so EASY to do • Fibrinogen • Factor Assays

RBC LAB http: //www. chronolab. com/hematology/2_1. htm
- Slides: 76