Randomised Controlled Trials RCTs Professor Fiona Alderdice School
 Professor Fiona Alderdice School of Nursing and Midwifery")







 Not randomised")




















- Slides: 31
Randomised Controlled Trials (RCTs) Professor Fiona Alderdice School of Nursing and Midwifery
What is a RCT • A prospective study comparing the effect and value of an intervention or interventions against a control in human participants • The intervention may be prophylactic, diagnostic or therapeutic • Persons in the intervention and control groups must be enrolled, treated and followed up over the exact same time frame
Types of study designs used in nursing research • Descriptive – Quantitative methods • • Surveys Case studies Case series Routine data – Qualitative methods • • • Participant observation Conversation/video analysis Text/discourse analysis Interviewing Focus Groups • Causation – RCTs – Cohort studies – Case control studies
Randomised Controlled Trials • What is the effect of this intervention? • Used with a controlled intervention of likely benefit • Primary method of studying new therapies
RCTs: Why and when? • “Gold Standard” method • To determine if a particular intervention has the anticipated effect, no effect or cause harm • Conducted because preliminary evidence suggests that the outcome will influence practice • Should be tested before a treatment becomes a part of routine care without proof of benefit • There is a ‘window of opportunity’ within which a trial can be conducted
Trial Designs • Drug Trials Phase I – IV • • Parallel group designs Cross over trial designs Cluster Trials Factorial
Drug Trial Phases Phase Who Design Outcomes I Healthy volunteers Not randomised Pharmacology and toxicology to establish a safe dose II Patients with the condition Not randomised Preliminary information on efficacy Additional safety information III Patients with the condition Randomised To assess dosage effects, Control efficacy and safety IV Patients with the condition Post marketing study Long term outcomes including safety and efficacy
Complex interventions • Many nursing and midwifery interventions are complex interventions. It is important to define the intervention clearly, identifying each component and how to administer the intervention in practice.
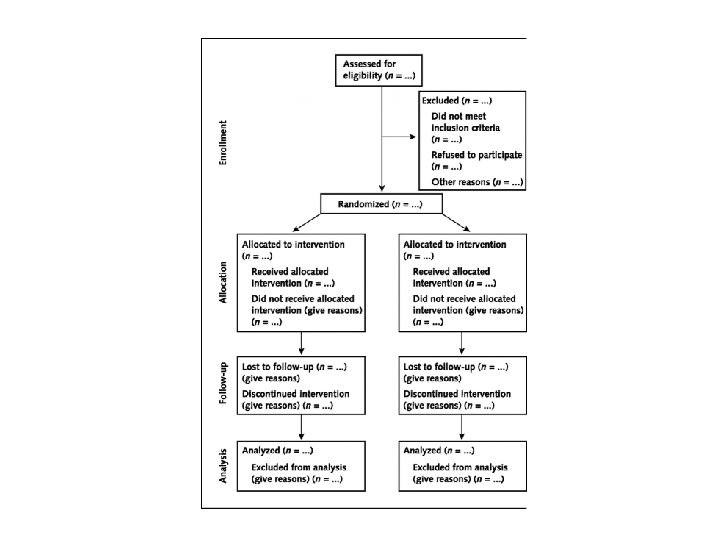
Parallel Group • Parallel Group Design Assessed for eligibility (n = 1283) Not randomised (n = 639) Not eligible (n = 289) Declined (n = 350) Randomised (n = 644) Control (n = 323) Example of a parallel group design Intervention (n = 321)
Cross-Over Design Washout Treatment B Treatment A Treatment B Assessed for eligibility Randomisation Week: 1 Example of a cross over design 8 14 22
Cluster Randomised Trials
2 X 2 Factorial Design
Example of a 2 X 2 factorial design Fiona Alderdice, Jenny Mc. Neill, Toby Lasserson, Elaine Beller, Margaret Carroll, Vanora Hundley, Judith Sunderl and, Declan Devane, Jane Noyes, Susan Key, Sarah Norris, Janine Wyn-Davies and Mike Clarke Do Cochrane summaries help student midwives understand the findings of Cochrane systematic reviews: the BRIEF randomised trial. Systematic Reviews 2016 http: //systematicreviewsjournal. biomedcentral. com/articles/10. 1186/s 13643 -016 -0214 -8
Study Population • A subset of the population of interest • Should be clearly defined in advance • Eligibility Criteria – Inclusion criteria • To ensure that the participants enrolled have the potential to benefit from the intervention • To ensure that it is likely that the hypothesised result is detected – Exclusion criteria • To ensure that no participants for whom the treatment is harmful are enrolled
Randomisation • Aims to avoid selection bias • Tends to guarantee comparability of treatment groups at study entry • Simple Randomisation – Similar to flipping a coin – Could be an imbalance at any one point • Block Randomisation
Blinding • Double-blind design is the ideal to avoid potential problems of bias in data collection and assessment (can also have triple and quadruple blinding!) • Single-blinded design • Non-blinded design
Blinding in randomised trials: hiding who got what (Shultz and Grimes Lancet 2002; 359: 696 -700 ) • • • Participants – Less likely to have biased psychological or physical responses to intervention – More likely to comply with trial regimens – Less likely to seek additional adjunct interventions – Less likely to leave trial without providing outcome data, leading to lost to follow-up Trial Investigators – Trial Less likely to transfer their inclinations or attitudes to participants – Less likely to differentially administer co-interventions Less likely to differentially adjust dose – Less likely to differentially withdraw participants – Less likely to differentially encourage or discourage participants to continue trial Assessors – Less likely to have biases affect their outcome assessments, especially with subjective outcomes of interest
Power and Sample Size • The number of participants required to answer the main trial question is known as the sample size • The sample size is calculated using a power calculation • A RCT should have significant power to detect differences between the groups
Power and Sample Size Sample size justification Assuming an inter-patient SD in Hb. A 1 c of 1. 4% (as observed in the DAPIT study population, n=505, unpublished data) a total sample size of 180 patients recruited gives in excess of 90% power to detect (P<0. 05; two-tailed) a 0. 7% difference in mean Hb. A 1 c between groups. Allowing for practice and patient attrition, a total sample size of 144 (72 per group) still gives 85% power to detect this same difference. standard deviation of the outcome Number per arm (group) n = 2. (za/2 + zb)2. (s/d)2 0. 05, 0. 01 or 0. 001 (typical values) 0. 20, 0. 15 or 0. 10 (typical values) largest difference that you would allow to go undetected
Issues in Data Analysis • Pre-specified analysis plan • Intention to treat analysis • Sub-group analysis
Pre-specified analysis plan • Prior to recruitment of any patients the outcomes that are going to be assessed should have been defined in the study protocol • Primary outcomes should have sufficient statistical power to detect differences between the groups • Other pre-specified outcomes are know as secondary outcomes
Intention to Treat Analysis • Analysis must be by the treatment assigned – Even if a patient withdraws from the study early or is a poor complier they should still be included in the analysis – To remove them undermines the randomisation process and may introduce bias
Subgroup Analysis • Should be defined by a factor assessed at baseline • Should be pre-specified in the analysis plan to reassure reader that investigator has not ‘gone fishing’ • Important, but remember there will be small numbers in subgroups, therefore will have lower power – interpret results with some caution.
Interpreting Results An oversimplification of the interpretation of p values and ratios Measure p values Result Comment p=0. 51 Not statistically significant p=0. 06 Not statistically significant (trend towards significance) p=0. 02 Statistically significant p<0. 05 Statistically significant p<0. 001 Highly Statistically significant 1. 35 (95% CI 1. 09 – 1. 67) Statistically significant (more likely to have event) 1. 18 (95% CI 0. 30 – 4. 57) Not statistically significant 0. 67 (95% CI 0. 25 – 1. 79) Not statistically significant 0. 51 (95% CI 0. 32 – 0. 82 ) Statistically significant (less likely to have event) Ratios
Reporting Clinical Trials • CONSORT 2010 Consolidated Standards of Reporting Trials • Trials should be registered and protocol and results papers should be reported according to the CONSORT guidelines
Principles of Good Trial Design • Random Assignment • Allocation Concealment – secure randomisation • Checking the validity of randomisation: are baseline prognostic variables similar? • Blinding – double, single • Eligibility criteria specified • Sample size calculation and assumptions reported • Compliance with treatment regimen described • Loss to follow-up rate quoted or calculable • Intention to treat analysis • Choice of suitable outcome measures • Primary outcome presented with confidence interval • Subgroup analysis pre-specified • Reported according to CONSORT Daniels & Hills (2006)
Additional references • CONSORT statement http: //www. consort-statement. org/ • Daniels J and Hills RK (2006) Design clinical trials in women’s health. Reviews in Gynaecological and Perinatal Practice 6 (2006) 33– 39 http: //www. qtnpr. ca/files/Designing%20 Clinical%20 Trials-Daniels. pdf Wolfgang Viechtbauer , Luc Smits, Daniel Kotz, Luc Budé, Mark Spigt, Jan Serroyen, Rik Crutzen A simple formula for the calculation of sample size in pilot studies Journal of Clinical Epidemiology, Vol. 68, Issue 11, p 1375– 1379 • Published online: June 5 2015 http: //www. jclinepi. com/article/S 08954356(15)00303 -0/pdf